Mechanisms of Osteoclastogenesis in Orthodontic Tooth Movement and Orthodontically Induced Tooth Root Resorption
Article information

Abstract
Orthodontic tooth movement (OTM) is achieved by the simultaneous activation of bone resorption by osteoclasts and bone formation by osteoblasts. When orthodontic forces are applied, osteoclast-mediated bone resorption occurs in the alveolar bone on the compression side, creating space for tooth movement. Therefore, controlling osteoclastogenesis is the fundamental tenet of orthodontic treatment. Orthodontic forces are sensed by osteoblast lineage cells such as periodontal ligament (PDL) cells and osteocytes. Of several cytokines produced by these cells, the most important cytokine promoting osteoclastogenesis is the receptor activator of nuclear factor-κB ligand (RANKL), which is mainly supplied by osteoblasts. Additionally, osteocytes embedded within the bone matrix, T lymphocytes in inflammatory conditions, and PDL cells produce RANKL. Besides RANKL, inflammatory cytokines, such as interleukin-1, tumor necrosis factor-α, and prostaglandin E2 promote osteoclastogenesis under OTM. On the downside, excessive osteoclastogenesis activation triggers orthodontically-induced external root resorption (ERR) through pro-osteoclastic inflammatory cytokines. Therefore, understanding the mechanisms of osteoclastogenesis during OTM is essential in reducing the adverse effects of orthodontic treatment. Here, we review the current concepts of the mechanisms underlying osteoclastogenesis in OTM and orthodontically induced ERR.
INTRODUCTION
Orthodontic tooth movement (OTM) is the process by which orthodontic force — mechanical force deliberately delivered by orthodontic appliances — causes the tooth to move within the alveolar bone due to an accelerated bone remodeling process. The tooth is attached to the alveolar bone through the periodontium, which consists of several units of mineralized and non-mineralized tissues, such as the cementum on the tooth root surface, the periodontal ligament (PDL), the alveolar bone, and the gingiva.[1] The PDL is a dense connective tissue that plays a critical role in supporting the tooth within its socket. It comprises various components, including bundles of collagenous fibers primarily composed of type I collagen. The Sharpey’s fibers are the terminal segment of the PDL that insert into the cementum and the alveolar bone on each side. They are aligned in different orientations across different locations to anchor the tooth to the surrounding alveolar bone. The cementum is a thin mineralized tissue layer covering the tooth root. The integrity of the cementum is important for tooth stability and protecting the tooth from external resorption.[2] The alveolar bone is a mineralized tissue that undergoes a finely controlled process, coordinating bone resorption by osteoclasts and bone formation by osteoblasts.[3] It has a high rate of bone turnover compared to the axial and appendicular skeleton.[4,5] One unique aspect of the alveolar bone is that it is of the neural crest origin.[6] Orthodontic force is transmitted from the tooth to the alveolar bone through the surrounding PDL, resulting in reversible micro-injuries of the PDL. This triggers the movement of the teeth within the alveolar bone. During the initial application of orthodontic force, the tooth moves within the PDL space, causing the PDL to either stretch or compress. On the tension side where PDL is stretched, osteoblastic activities are enhanced, which results in osteoid deposition, mineralization, and eventually new bone formation. On the compression side, bone resorption occurs through the activation of osteoclasts via a sterile inflammatory response triggered by proinflammatory cytokines.[7] These proinflammatory cytokines contribute to bone resorption by inducing the expression of a receptor activator of nuclear factor (NF)-κB ligand (RANKL), a member of the tumor necrosis factor (TNF) family. RANKL binds to its receptor RANK to induce osteoclastogenesis.[8,9] Excessive compressive force generated by orthodontic force can also stimulate a significant increase in RANKL expression in periodontal tissues, potentially leading to the pathological resorption of the cementum termed orthodontically induced external root resorption (ERR). ERR is irreversible when the resorption involves the dentin.[10,11]
Malocclusion is one of the most common dentoalveolar anomalies that impair oral function and craniofacial growth, typically associated with the impairment in breathing, chewing, speaking, and facial appearance. Therefore, the demand for orthodontic treatment is strong. However, orthodontic treatment is a lengthy process, typically spanning over 2 years with an inherent risk for tooth root resorption. Therefore, it is crucial to have better understanding of the fundamental biological mechanisms of OTM and orthodontically induced ERR to ameliorate risks associated with orthodontic treatment. This review summarizes the current concept of the mechanisms of osteoclastogenesis during OTM and orthodontically induced ERR.
CYTOKINES THAT PROMOTE OSTEOCLASTOGENESIS DURING OTM
1. RANKL/RANK/OPG
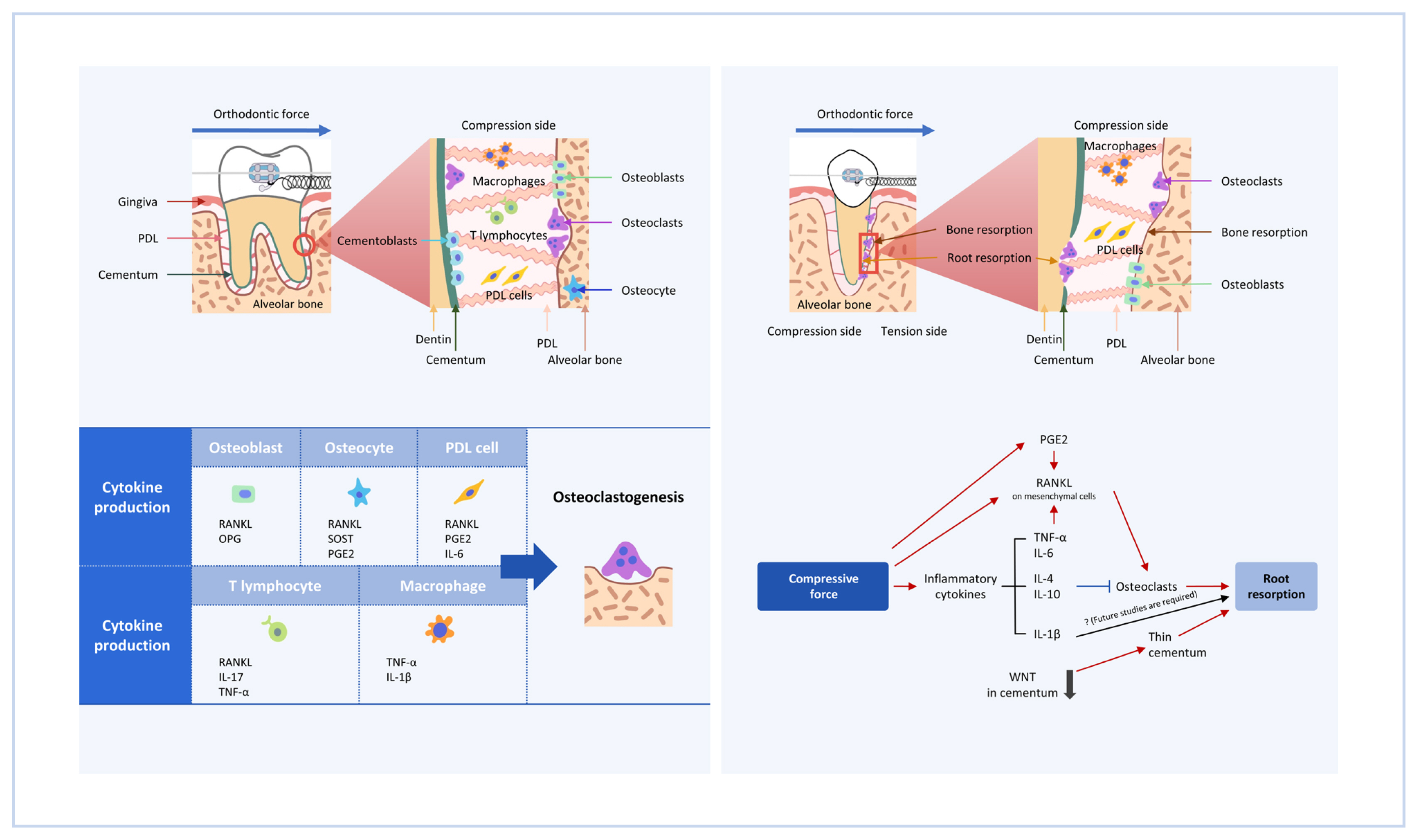
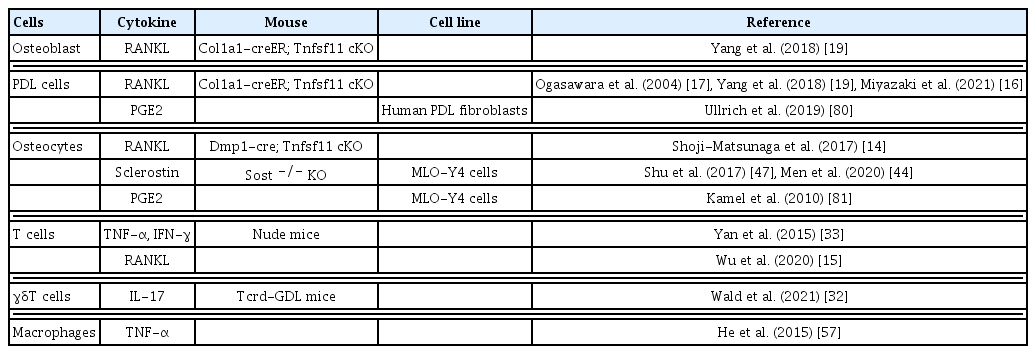
RANKL, a member of the TNF family, is an essential cytokine for osteoclastogenesis.[8,9] RANKL binds to its receptor RANK, which is expressed on osteoclast precursor cells, to induce osteoclastogenesis through the activation of transcription factors, such as the NF of activated T cells 1 (NFATc1).[12] The importance of RANKL in bone metabolism in vivo has been demonstrated by studies of RANKL-knockout (KO) mice. RANKL- and RANK-KO mice show osteopetrosis with a complete absence of osteoclasts.[9] In addition, RANKL-KO mice are protected from bone erosion during arthritis, demonstrating that osteoclastogenesis under inflammatory conditions is dependent on RANKL.[13] Among the cells constituting the periodontal tissues, it has been reported that RANKL is expressed by PDL cells, osteoblasts, osteocytes, and T lymphocytes.[3,14–17] The RANKL-RANK pathway critically contributes to OTM, as many osteoclasts are observed on the compression side of the tooth in a sterile inflammatory condition induced by orthodontic forces. In fact, the administration of anti-RANKL antibodies delays OTM.[14] Therefore, efforts have been made to elucidate which cellular source of RANKL in periodontal tissues is important for OTM (Fig. 1, Table 1).

Schematic diagram of orthodontic tooth movement (OTM). OTM is induced by applying orthodontic force to a tooth. On the side of the alveolar bone upon which the tooth root is compressed against (“compression side”), osteoclasts are formed to resorb the alveolar bone, to create a space to which the tooth root moves. Cytokines produced by several cell types, including osteoblasts, osteocytes, periodontal ligament (PDL) cells, lymphocytes and macrophages play roles in inducing osteoclastogenesis. RANKL, receptor activator of nuclear factor-κB ligand; OPG, osteoprotegerin; SOST, sclerostin; PGE2, prostaglandin E2; IL, interleukin; TNF, tumor necrosis factor.
Cytokine-producing cells that induce osteoclastogenesis during orthodontic tooth movement
One clinically important observation is that temporary anchorage devices, which are widely used as an orthodontic anchorage, or ankylosed teeth, which are fused to alveolar bones, don’t move even with the persistent application of orthodontic force.[18] This has led to the theory that PDL cells may serve as a primary source of RANKL in OTM. Ogasawara et al. [17] show RANKL expression in periodontal tissues by in situ hybridization, reporting that osteoblasts and PDL cells express Tnfsf11 (encoding RANKL) mRNAs under physiological conditions. During OTM, RANKL-positive PDL cells increase on the compression side. Functionally, RANKL deletion in osteoblasts and PDL cells using a tamoxifen-inducible 3.2 kb collagen1α1 promoter creER (3.2 kb Col1a1-creER) transgenic line blocks OTM.[19] Therefore, osteoblasts and PDL cells are an important source of RANKL in OTM.
Factors supporting the formation of PDL are involved in regulating RANKL expression. The transcription factor mohawk homeobox (Mkx) plays an important role in tendon maturation by regulating the expression of type I collagen. Mkx is also expressed in the PDL and is responsible for periodontal tissue homeostasis by suppressing the differentiation of PDL cells into osteoblasts.[20] In Mkx-KO rats, OTM was suppressed through a decline in RANKL expression in PDL cells.[16] Another important molecule, periostin, is a secreted extracellular matrix protein highly expressed in PDL and periosteum, regulating periodontal tissue homeostasis. Periostin-KO mice showed a reduced number of RANKL-positive stromal cells and osteoclasts on the compression side, consequently inhibiting OTM.[21]
Osteocytes account for about 90% of the cells that make up bone and are by far the largest in number among bone-constituent cells. Two comprehensive genetic engineering studies show that RANKL deletion by Dmp1-cre, which marks a majority of osteocytes, reduces the number of osteoclasts during the adult stage.[22,23] These studies show the primary source of RANKL under physiological conditions in adult stages is predominantly osteocytes.
Osteocytes extend their dendrite-like projections in the bone matrix and are in close contact with other osteocytes. This osteocyte network is thought to regulate bone homeostasis by enabling the sensing and response to mechanical stimuli transmitted through bone.[24,25] Therefore, it is postulated that osteocytes respond to orthodontic forces and are involved in OTM. In the osteocyte-ablated model using the diphtheria toxin receptor-mediated cell ablation system under the control of a Dmp1 promoter, there is a decrease in the number of osteoclasts on the compression side and a delay in OTM.[26] Additionally, Shoji-Matsunaga et al. [14] demonstrate that the number of osteoclasts on the compression side are reduced when OTM is induced in mice in which RANKL is ablated by Dmp1-cre. However, Dmp1-cre has been shown to be active in cell types other than osteocytes.[27–29] Therefore, experiments with a truly osteocyte-specific cre line is needed to definitively support the above-described osteocyte-centric notion on OTM. More specifically, the source of RANKL in OTM should be clarified in future studies.
Surgical techniques, such as corticotomy, are widely applied in orthodontic practice to accelerate orthodontic treatment.[30] It is speculated that this intervention induces an accumulation of immune cells due to corticotomy-induced bone damage, which speeds up OTM.[31] Immune cells located in the gingiva and PDL contribute to osteoclastogenesis.[32] For example, OTM is delayed in nude mice lacking mature T cells.[33] When T cells are transplanted into these mice, OTM is accelerated associated with increased accumulation of T cells on the compression side. Interestingly, it is reported that RANKL-positive activated T cells increase in mice transplanted with CD4 positive T cells.[15] Transplantation of T cells also increases the number of TNF-α and interferon-γ (INF-γ) positive cells on the compression side, indicating that T cells are one of the sources of inflammatory cytokines. Furthermore, Wald et al. [32] show that γδT cells, which are innate-like lymphocytes, contribute to the acceleration of OTM. This is demonstrated using Tcrd-GDL mice, allowing the conditional ablation of γδT cells under administration of diphtheria toxin, in which OTM is inhibited. They also report that a greater proportion of γδT cells reside in the gingiva, thereby proving that γδT cells contribute to OTM. γδT cells in PDL are mainly interleukin (IL)-17 producing Vγ6 positive γδT cells with an elevated expression of IL-17 during OTM. IL-17 has been reported to induce RANKL expression in PDL cells.[34] Taken together, these reports indicate that immune cells play a pivotal role in OTM through the RANKL-RANK pathway.
Osteoprotegerin (OPG) is a member of the TNF receptor family and plays a significant role in regulating osteogenesis by acting as a decoy receptor for RANKL, inhibiting RANKL-RANK signaling.[35] The regulation of bone remodeling depends on achieving a balance between OPG production and RANKL-RANK binding. The RANKL/OPG ratio is a determining factor in the formation of osteoclasts. In OPG-KO mice, osteopenia develops due to enhanced osteoclastogenesis.[36] Conversely, mice with OPG overexpression show a notable suppression of bone resorption, resulting in severe osteopetrosis.[35] The main source of OPG production in bone tissue is osteoblasts.[37] Clinically, OPG concentration in gingival crevicular fluid (GCF) significantly decreases after orthodontic force application.[38] In the compression side of OTM, bone resorption is facilitated by an up-regulation of RANKL and a down-regulation of OPG.[38,39] Local injection of OPG-Fc results in decreased OTM due to the inhibition of osteoclastogenesis.[40,41] From these findings, OPG is a decoy receptor against RANKL, and is involved in bone remodeling through the RANKL/RANK/OPG system.
2. Sclerostin (SOST)
SOST is a negative regulator of bone formation, which is encoded by the Sost gene and is primarily expressed by mature osteocytes. SOST binds to lipoprotein related peptide (LRP)5, LRP6, and frizzled receptors. It antagonizes osteoblast differentiation via inhibiting the Wnt/β-catenin signaling pathway. Deficiency in SOST gene results in high bone mass genetic disorders such as sclerosteosis.[42] Osteocytes translate mechanical loading into SOST production. For example, SOST expression is increased in unloaded sites in a tail suspension experiment.[43] In periodontal tissues, osteocytes in the alveolar bone express SOST, as revealed by Sost-LacZ mice.[44] During OTM, SOST is significantly increased on the compression side of the alveolar bone. One study postulates that when orthodontic force is initiated, bone cells of the compression side are unloaded, thereby increasing SOST expression.[45] By injecting SOST protein locally at the compression side of the alveolar bone, osteoclastogenesis is promoted in association with accelerated OTM.[46] It is also reported that osteoclastic activities and RANKL expression are reduced on the compression side in SOST-KO mice, resulting in a slower OTM rate. This indicates that osteoclastic activity and RANKL expression are decreased due to the lack of SOST.[47] SOST affects not only osteoclastogenesis but also osteogenesis. It is postulated that mesenchymal stem cells reside in the PDL and give rise to PDL cells and osteoblasts in the alveolar bone.[48] Gli1-positive cells in PDL include a population of mesenchymal stem cells termed PDL stem cells (PDLSCs). SOST suppresses PDLSCs activities by inhibiting the Wnt/β-catenin signaling pathway.[44] As a result, osteogenic capacity by PDLSCs is decreased, creating space for tooth movement on the compression side. Therefore, SOST is an important cytokine that can potentially accelerate OTM.
3. Inflammatory cytokines in OTM
One aspect of orthodontic treatment is that it induces an aseptic inflammatory condition. OTM has been reported to increase inflammatory cytokines, such as TNF-α, IL-1, IL-6, and IFN-γ [49–51] (Fig. 1, Table 1). Since these cytokines are involved in osteoclastogenesis directly as well as indirectly through modulating inflammatory responses, it is important to understand how inflammatory cytokines regulate OTM.
TNF-α is expressed by activated macrophages, T and B lymphocytes, and natural killer cells.[52] Excessive activation of TNF-α signaling is associated with chronic inflammation. Mechanistically, TNF-α directly binds to receptors on the surface of osteoclasts and promotes osteoclastogenesis in a manner dependent on the RANKL/RANK axis.[53–55] Also, TNF-α stimulates RANK expression in osteoclast precursor cells.[56]
Administration of TNF-α inhibitor, etanercept, suppresses OTM.[33,57] Adoptive transfer experiments with T cells reveal that TNF-α positive cells accumulate on the compression side.[33] Similarity, M1 macrophages, known to produce inflammatory cytokines, increase the number of TNF-α positive cells, as shown by adoptive M1 macrophage transfer experiments.[57] These results indicate that TNF-α producing cells are primarily T cells and macrophages during OTM. Recently, an indirect mechanism of TNF-α in OTM has been reported. Ohori et al. [58] report that OTM is suppressed in TNF receptor (TNFR) 1 and TNFR2 double KO (dKO) mice. A decrease in SOST-positive osteocytes on the compression side is observed in TNFR dKO mice. This observation shows that TNF-α accelerates OTM by inducing SOST expression. In conclusion, TNF-α positively regulates OTM.
IL-1 regulates adaptive and innate immunity by binding to the IL-1 type 1 receptor (IL-1R). IL-1 is synthesized primarily by monocytes but also by activated macrophages, granulocytes, and endothelial cells.[59,60] The 2 forms of IL-1, IL-1α, and IL-1β have similar biological activities because of their common receptors on target cells. In addition to the immune response, IL-1 also promotes osteoclastogenesis. In vitro experiments show that the IL-1 administration upregulates RANKL expression in osteoblasts and promotes osteoclastogenesis.[61,62] IL-1α KO, IL-1β KO, and IL1-α/β dKO mice exhibited an increase of bone mass associated with a decrease in the number of osteoclasts.[63] These observations clearly indicate that excessive IL-1 signaling under pathological conditions enhances bone resorption. IL-1β is expressed on the compression side in OTM.[64] Additionally, IL-1R inhibitors reduce OTM associated with the decrease of osteoclasts.[65]
IL-6 is a cytokine with a pleiotropic effect on hematopoiesis, inflammation, immune responses, and bone homeostasis.[66] IL-6 is produced by various types of cells including immune cells, endothelial cells, adipocytes, and PDL cells. IL-6 is upregulated under inflammatory conditions such as periodontitis, rheumatoid arthritis and OTM.[67–69] Tsukasaki et al. [70] show that PDL cells highly express Il6 mRNA during periodontitis by in situ hybridization, indicating that PDL cells may act as a source of IL-6 in OTM. IL-6 binds to IL-6R and gp130 on osteoblastic cells. It activates the signal transducer and activator of transcription 3 (STAT3) activated in the cytoplasm by Janus kinase 2 (JAK2), inducing RANKL expression.[71,72] In osteoblastic deletion of Stat3 by Col1a1-CreER mice, OTM is inhibited due to the reduction in osteoblasts associated with a decrease in the number of osteoclasts on the compression side.[73] In experiments with a co-culture system of osteoblasts and osteoclasts under mechanical stimulation, Stat3-deficient osteoblasts show a lower osteoclast induction ability. These results may suggest that mechanical stimulation by orthodontic treatment affects osteoclastogenesis by upregulating RANKL expression in osteoblasts via the IL-6-induced JAK2/STAT3 signaling pathway.
IFN-γ is a cytokine that is critical for innate and adaptive immunity against viral and some bacterial infection. IFN-γ induces major histocompatibility complex (MHC) class II on antigen-presenting cells and stimulates macrophages and dendritic cells to phagocytose and kill bacteria.[74] Dual roles of IFN-γ in osteoclastogenesis have been reported. In vitro experiments reveal that the presence of IFN-γ strongly suppresses osteoclastogenesis.[75] On the other hand, IFN-γ also activates CD4+ T cells to produce RANKL and TNF-α through enhanced MHC class II expression on antigen-presenting cells.[76] In OTM, IFN-γ positive cells are accumulated on the compression side.[33,77] As the number of IFN-γ positive cells are decreased in nude mice, T cells are one of the cells that produce IFN-γ during OTM.[33] Yan et al. [33] show no significant changes in OTM after blockage of IFN-γ. On the other hand, Kohara et al. [78] report that IFN-γ injection decreases OTM by reducing the osteoclast number on the compression side. These results support the notion that IFN-γ acts via positive and negative pathways during OTM.
4. Prostaglandin E2 (PGE2)
Most widely used medications in orthodontics are acetaminophen or nonsteroidal anti-inflammatory drugs (NSAIDs) for control of pain following mechanical force application to the tooth. The use of NSAIDs reduces PGE2 by inhibiting 2 cyclooxygenase enzymes. When orthodontic force is applied, arachidonic acid is released from the lipid bilayer due to disruption of the cells on the pressure side. PGE2 is a well-known inflammatory mediator produced from arachidonic acid by constitutively expressed cyclooxygenase (COX)-1 and cytokine-inducible COX-2. COX-1 is mostly synthesized in normal tissues, while COX-2 is induced in inflammatory site.[79] Clinically, elevated PGE2 in the GCF is observed in patients undergoing orthodontic treatment.[49] This phenomenon occurs due to the PGE2 production by osteocytes and PDL cells in response to orthodontic forces.[80,81] From the 1980s, some orthodontists advocated that PGE2 accelerates OTM, since PGE2 acts directly on osteoclastogenesis.[82] The actions of PGE2 in the target cells are mediated by 4 different G protein-coupled receptor subtypes which are EP1, EP2, EP3, and EP4. Among PGE2 receptor subtypes, EP4 has been shown to mainly mediate PGE2-induced RANKL expression in osteoblasts.[82,83] OTM is accelerated in mice injected with PGE2.[84] These findings indicate that PGE2 produced by orthodontic forces promotes OTM.
ORTHODONTICALLY INDUCED ERR
ERR occurs in various conditions, such as bacterial infection, trauma, and orthodontic treatment. Histologic studies reported over 90% of patients undergoing orthodontic treatment have some evidence of ERR.[85] The tooth root is protected from occlusal and orthodontic forces by the cementum. External dentin resorption would not occur unless full-thickness cemental resorption.[86] Proliferation and differentiation of cementoblasts are regulated by Wnt/β-catenin signaling. Loss of Wnt/β-catenin signaling in odontoblasts and osteoblasts by Ocn-cre induces a thinner cementum.[87] In other words, the reduction of canonical Wnt signaling negatively affects cementogenesis, resulting in a thinner cementum. This causes ERR without the application of orthodontic force, indicating cementoblasts are an important tissue that prevents ERR. In addition, ERR also occurs in the presence of cementum resorption by osteoclasts. Thus, pro-osteoclastic cytokines such as RANKL, TNF-α, and PGE2 facilitate ERR. M1 macrophages produce pro-inflammatory factors such as IL-6, TNF-α, and IL-1β, whereas M2 macrophages produce IL-4 and IL-10 which are involved in anti-inflammatory responses. He et al. [88] propose the M1/M2 ratio as one indicator of the progression of orthodontic induced ERR. Following the application of orthodontic force, an accumulation of CD68+iNOS+ M1 macrophages is observed at the ERR site. On the other hand, when orthodontic forces are removed, CD68+CD163+ M2-like macrophages increase. When the M1/M2 ratio is reduced by administration of IL-4, ERR is decreased. Fang et al. [89] also report that the inhibition of the CXCL12-CXCR4 axis with AMD 3100 suppresses the migration of M1 macrophages to the ERR sites and prevents orthodontic-induced ERR.
Several cytokines involved in RANKL expression induce ERR during OTM. Notch signaling pathway regulates cell growth, cell death, and differentiation programs via cell-cell communication. In the skeleton, 4 Notch receptors (Notch1 to −4) and 5 ligands (Delta1, −3, and −4 and Jagged [JAG]1 and −2) are expressed.[90] The JAG1-Notch2 signaling pathway induces RANKL expression on osteoblasts. Kikuta et al. [91] report that an increase in JAG1 and RANKL-positive cells around the ERR sites are observed during OTM. In vitro experiments indicate mechanical stimulation of human PDL (hPDL) cells increases JAG1 expression. The inhibition of Notch signaling by a selective γ-secretase inhibitor suppresses RANKL in hPDL cells.
Another cytokine that upregulates RANKL expression in periodontal tissues is parathyroid hormone (PTH)-related protein (PTHrP). PTHrP is a member of the PTH family secreted by defined skeletal cell types, including resting zone chondrocytes that include skeletal stem cells. During endochondral bone formation, PTHrP inhibits proliferating chondrocyte differentiation. In addition, PTHrP is involved in tooth eruption.[92,93] Dental follicle progenitor cells and cells around the root surface express PTHrP, and these cells contribute to tooth eruption via autocrine PTHrP-PTH1R signaling.[94] PTHrP induces RANKL expression in PDL cells.[95] RANKL on PDL cells has been reported to be involved in root resorption in deciduous teeth, thus PTHrP may be involved in ERR.[96]
Cytokines that promote osteoclastogenesis cause ERR, whereas the effect of IL-1β on OTM is controversial. Clinically, polymorphism at IL-1β genes which reduces the IL-1β cytokine production have higher risks to experience ERR.[97] P2X purinergic receptor 7 (P2X7) is an adenosine triphosphate (ATP)-gated ionotropic channel. It can be activated (opened) after binding to extracellular ATP, a danger signal from cells under mechanical stimuli. Opening the channel causes the accumulation of intracellular calcium and releases IL-1β. Viecilli et al. [98] reported that advanced ERR was observed in P2X7 KO mice during OTM. Similarly, OTM in IL-1β KO mice showed progression of ERR compared to wild-type mice.[99] On the other hand, IL-1β was originally discovered as a cytokine that induces bone degradation, and the positive effects on osteoclastogenesis are well established.[61–63,100] Furthermore, IL-1β and compressive forces lead to a significant induction of RANKL expression in primary human cementoblasts, indicating that the IL-1β production leads to ERR during OTM.[101] Therefore, future studies are required to elucidate the detailed molecular mechanisms by which IL-1β affects ERR.
As mentioned earlier, RANKL-positive cells promote ERR. RANKL is primarily produced as a membrane protein. Some membrane-bound RANKL is then shed by matrix metalloproteinases (MMPs) such as MMP13 and ADAM10 to form soluble RANKL.[102] Previous studies have reported that membrane-bound RANKL is a more potent inducer of osteoclastogenesis.[102,103] Soluble RANKL doesn’t contribute to osteoclastogenesis in young mice and OVX induced bone loss.[104,105] However, it is unclear whether soluble RANKL contributes to ERR. If soluble RANKL is not involved in ERR, osteoclasts that cause root resorption receive RANKL signaling through cell-cell contact around root resorption sites. This may establish a new treatment approach that suppresses ERR without affecting OTM by selectively inhibiting membrane-bound RANKL around root resorption areas.
In conclusion, cytokines that activate osteoclastogenesis, except IL-1β promote ERR (Fig. 2).
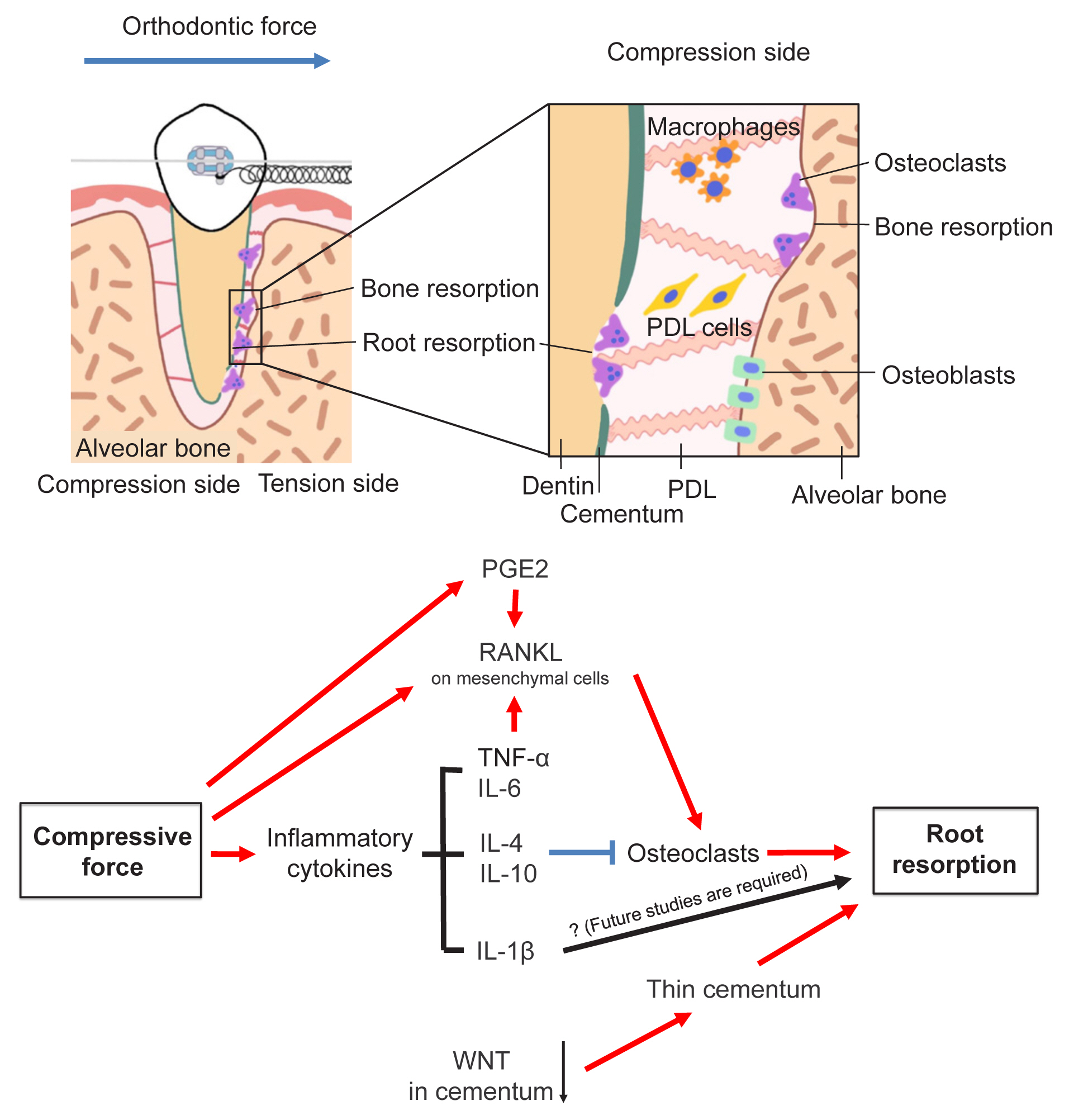
Schematic diagram of orthodontically induced external root resorption (ERR). Excessive orthodontic forces can induce a pathological condition termed orthodontically induced ERR, which is characterized by the resorption of the tooth root in addition to the alveolar bone. Cytokines that are involved in normal orthodontic tooth movement are also involved with tooth root resorption. PDL, periodontal ligament; PGE2, prostaglandin E2; RANKL, receptor activator of nuclear factor-κB ligand; TNF, tumor necrosis factor; IL, interleukin.
CONCLUSION AND PROSPECTS
During OTM, bone formation by osteoblasts and bone resorption by osteoclasts are tightly regulated. In addition, recent studies implicate the importance of additional cell types, including osteocytes, immune cells and PDL cells for osteoclastogenesis. Complex signaling mediated by these cells causes osteoclastogenesis, making it difficult to comprehensively map cell-to-cell signaling networks. With the development of genetic engineering technology, it has become easier to analyze the function of specific cells for the purpose of elucidating the molecular mechanisms underlying OTM and ERR. This knowledge is expected to be clinically applicable to orthodontic treatment in the future. IL-1β acts positively on OTM, but its role in orthodontic-induced ERR is not fully understood. If IL-1β is proven to promote OTM without causing ERR, it would be one of the potential therapeutic targets.
Loss of Wnt/β-catenin signaling causes ERR under physiological conditions. Additionally, other genes involved in ERR have been reported.[106,107] However, only a limited number of studies have analyzed the function of these genes during OTM. Elucidating how these genes are involved in OTM is expected to pave the way for new orthodontic treatment modalities with fewer side effects.
Notes
Funding
This work was supported by grants from the National Institutes of Health (R01DE030630 to N.O., and R01DE029181, R01DE030416 to W.O.).
Ethics approval and consent to participate
Not applicable.
Conflict of interest
No potential conflict of interest relevant to this article was reported.