INTRODUCTION
Vitamin D (VD) is a fat-soluble vitamin, and it serves as one of the major hormones supporting bone health and calcium homeostasis.[1-4] Nutritional VD deficiencies can cause rickets in higher mammals during growth stages, and it can lead to osteomalacia in adults.[5] In addition, hereditary rickets can be caused by the genetic impairment of VD processing pathways or related signaling pathways. [6,7]
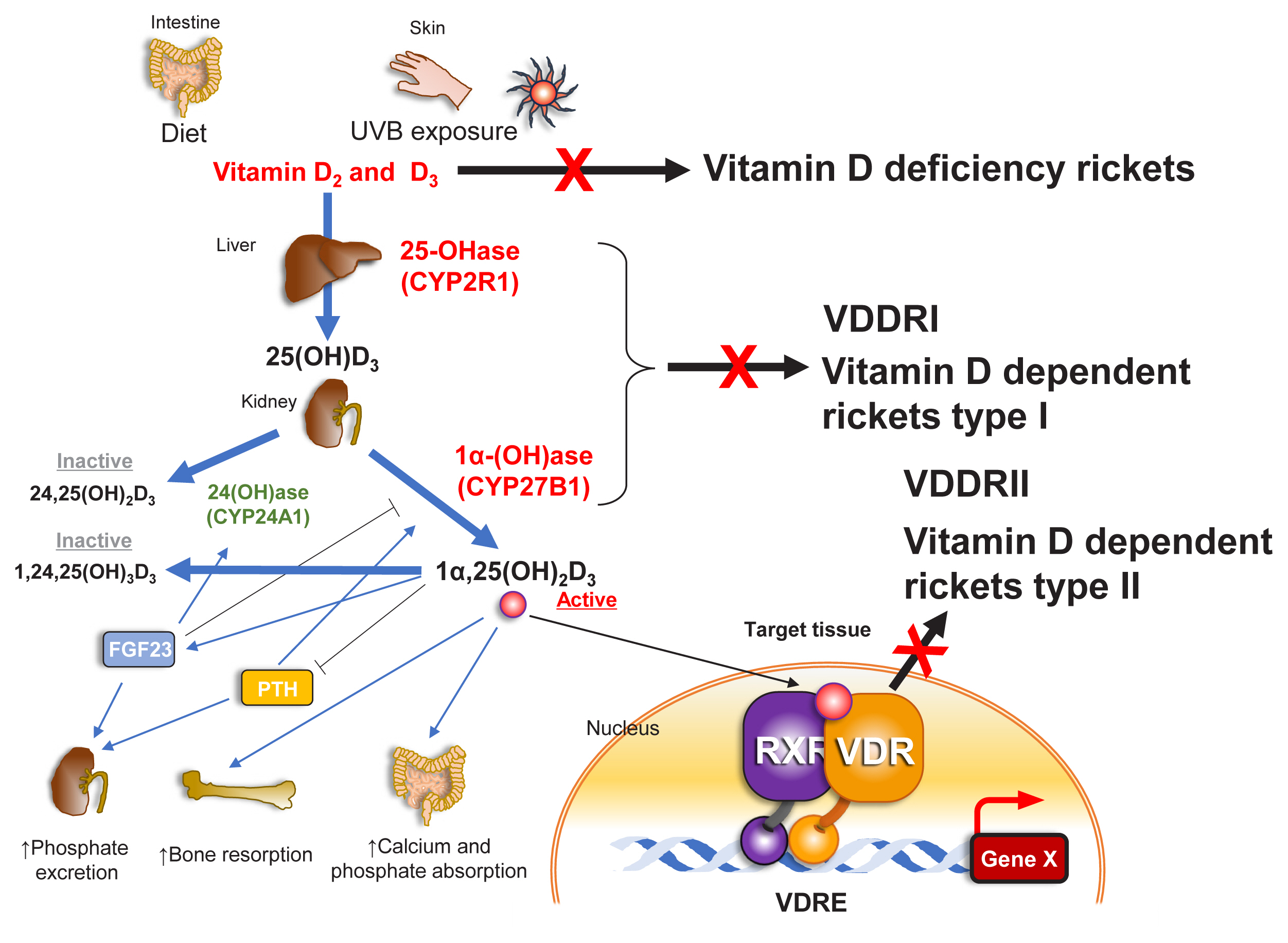
While genetic impairments of VD-associated proteins often lead to some generally similar outcomes, subtle differences in the symptoms caused by different genetic alterations can provide insight into the biological aspects of VD. For example, 2 types of hereditary rickets both cause bone disorders, but they differentially impact the skin. Type 1 hereditary rickets is caused by decreased activity of cytochrome P450 (CYP) 27B1, a key enzyme of VD biosynthesis that converts a VD precursor (25-hydroxy-VD3 [25(OH)D3]) into an active form of VD, 1α,25-dihydroxy-VD3 (1α,25[OH]2D3). [6] Type 2 hereditary rickets is caused by a malfunction of the VD receptor (VDR). Patients with type 2 hereditary rickets exhibit an identical rachitic abnormality to those with type 1 hereditary rickets, but type 2 is also characterized by alopecia, which is undetectable in type 1 patients (Fig. 1). These forms of hereditary rickets have been experimentally recapitulated by introducing null mutations into the mouse genes encoding CYP27B1 and VDR, and alopecia is observed only in mice that are genetically deficient in VDR. [4,6] This difference in outcomes between subjects lacking active VD and those lacking active VDR provides further evidence of the complexity of VD signaling and that it has tissue-specific effects.
In addition to its involvement in calcium metabolism, a wide variety of other biological events, including inflammation and cell proliferation, appear to be facilitated by VD.[7,8] Moreover, the dietary intake of sufficient VD is considered to slow the onset and development of many non-communicable chronic diseases, and multiple epidemiological studies have demonstrated correlations of the risks of particular disease events with nutritive VD deficiencies.[9,10] Unfortunately, dietary VD deficiencies are relatively common worldwide, even in some people with optimal diets.[2,11] As many disorders, including cancer, [12,13] have been related to inflammatory events, VD has been assumed to attenuate the inflammation process, though that hypothesis remains confirmed.[14-17] Given the fact that dietary VD deficiency is quite common globally, particularly in older adults, [2,11] an adequate intake of VD from diet and supplementation has been recommended to improve bone health and overall quality of life. [7,8] Additionally, VD and related pathways have been targeted in multiple therapeutic strategies.
BENEFICIAL ACTIONS OF VD IN EXTRA-SKELETAL EVENTS
As VD deficiency is considered to be a risk factor for the development of cancer, multiple clinical trials of therapies involving 1α,25(OH)2D3 or synthetic VD analogues have been conducted in the context of a variety of cancer types.[18,19] While treatments with 1α,25(OH)2D3 and synthetic VD analogues attenuate the proliferation of cultured cancer cell lines and the growth of xenografted tumors derived from certain types of human cancer cell lines, they have not yet been proven to inhibit the onset or development of cancers in human patients.[18-20]
The occurrence of several other non-communicable chronic diseases, including type 2 diabetes and hypertension, has been connected in some studies to the nutritional status of VD, though epidemiological outcomes have not been consistent among the reports.[9,10] However, studies of other disorders have suggested that VD alleviates inflammation in the context of multiple diseases and pathological events.[14,15] Despite these connections, similar to the clinical failure of VD administration to cancer patients, the clinical effects of VD supplementation and its analogues in other diseases remain to be established.
The lack of effectiveness of VD in clinical applications might be due in part to the relatively low doses of VD and synthetic analogues that can be administered.[21,22] Because these compounds are calcemic, over-supplementation can lead to hypercalcemia, and doses that are sufficiently high to attenuate disease development are thus not clinically applicable. Though the calcemic activities of VDR ligands are indispensable for bone health, the clinical potential of VDR ligands that specifically exert anti-inflammatory action would be desirable, and the development of such a new generation of VDR ligands is eagerly anticipated.
MECHANISMS ENSURING ADEQUATE SERUM VD LEVELS
VD is derived from dietary sources as a fat-soluble vitamin, but it is also produced endogenously from VD precursors.[6,23] The active form of VD, 1α,25(OH)2D3, is generated from its precursor (VD3) through 2 steps of hydroxylation, which are mainly catalyzed by hepatic CYP2R1 and renal CYP27B1. VD3 is first hydroxylated by CYP2R1 to form 25(OH)D3, which is subsequently converted into 1α,25(OH)2D3 by CYP27B1. Since CYP27B1 expression in the renal proximal tubule is highly sensitive and reciprocally regulated by parathyroid hormone (positive) and 1α,25(OH)2D3 (negative), the production of 1α,25(OH)2D3 is normally optimally balanced to meet bodily demands for serum VD.[6,23] When serum 1α,25(OH)2D3 is in excess, the activity of CYP27B1 is reduced, and 25(OH)D3 is hydroxylated by CYP24A1 in the proximal tubules to form 24,25(OH)2D3, which cannot be directly converted to the active form.[5,6]
Reflecting the significance of CYP27B1 and CYP24A1 in the tight regulation of the serum levels of 1α,25(OH)2D3, genetic inactivation of the gene encoding CYP27B1 causes type 1 hereditary rickets. In addition, inactivating genetic mutations of the gene encoding CYP24A1 were initially assumed to underlie the clinical symptoms of idiopathic infantile hypercalcemia.[24,25] Upon genomic sequencing of 5 patients, 5 genetic mutations were indeed identified in regions of the CYP24A1 gene that are important for the catalytic activity of the enzyme, and all 5 of the CYP24A1 mutant proteins were shown to be impaired in hydroxylation of 25(OH)D3. More recently, similar genetic mutations in CYP24A1 were found in adults, and these cases have suggested that inactivating mutations of this gene lead to hypercalcemia.[26] Mild inactivation of the CYP24A1 gene by single nucleotide polymorphisms or other genetic mutations has been discussed as a risk factor for the development of hypercalcemia in certain pathological settings.
FUNCTIONS OF VDR IN THE REGULATION OF GENE EXPRESSION
VDR, a member of the nuclear steroid hormone receptor superfamily, mediates the intracellular signaling of VD. Nuclear receptors (NRs), including VDR, act as ligand-dependent transcription factors that interact with response elements in the promoters of target genes, which include protein-coding genes as well as genes that code for non-translated RNA.[26,27] In the absence of VD, VDR is localized in the cytosol, but in the presence of 1α,25(OH)2D3, VDR undergoes nuclear translocation. In order to stably associate with DNA at a VD response element (VDRE), VDR heterodimerizes with a retinoid X receptor (RXR; α, β, γ); multiple genome-wide ChIP-sequencing studies have confirmed co-localization of VDR with RXRs on the chromatin of associated sites.
The consensus VDRE is composed of 2 motifs, with the sequence 5′-AGGTCA, separated by 3 bp.[3,26,28] Even though the VDR-RXR heterodimer binds to VDREs, the transactivation function of VDR is silent in the absence of 1α,25(OH)2D3. The binding of 1α,25(OH)2D3 induces the ability of the VDR-RXR heterodimer to activate gene transcription.[29]
TRANSCRIPTIONAL CO-REGULATORS IN GENE REGULATION BY VDR
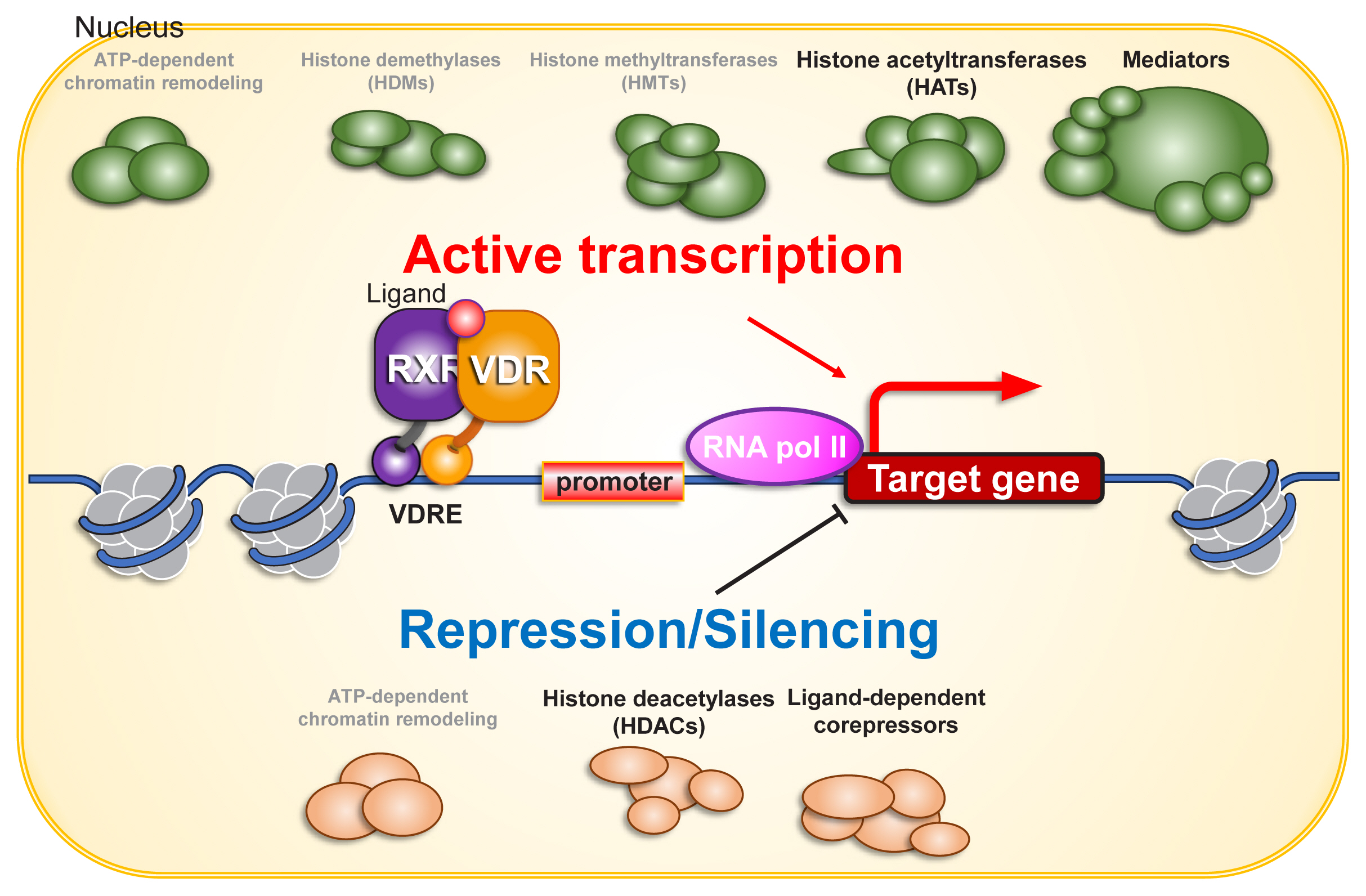
A number of transcriptional co-regulators, in addition to basal transcription factors, are involved in ligand-dependent activation of RNA polymerase II by VDR.[30,31] When transcriptionally silent unliganded VDR is bound to a VDRE, transcriptional co-repressors associate with VDR. Upon ligand binding, the co-repressors dissociate, and co-activators are recruited (Fig. 2).
Histone acetyltransferases (HATs) and histone deacetylases (HDACs) have been extensively characterized as co-regulators of the transcriptional activity of NRs.[30] Histone acetylation facilitates the transcriptional process by relaxing the nucleosome array and providing binding sites for proteins that associate with acetylated lysine. Accordingly, HATs act as co-activators for VDR and other NRs, while HDACs act as co-repressors. In addition, given the significance of methylation of lysine 4 and lysine 9 of histone H3, methyltransferases and demethylases that act on these 2 amino acids also influence the gene regulatory function of many NRs,[32,33] but they appear dispensable for gene regulation by VDR.
Among the NRs, sex hormone receptors interact with chromatin remodeling complexes, and chromatin reorganization influences the transcriptional regulation of these receptors.[34,35] However, VDR is unlikely to associate with chromatin remodeling complexes in the process of ligand-dependent gene regulation in most VD target cells. For example, one study employed Assay for Transposase-Accessible Chromatin sequencing (ATAC-seq) in the human keratinocyte cell line HaCaT and observed that chromatin organization is unaffected by the presence or absence of either 1α,25(OH)2D3 or VDR. Thus, the major transcriptional co-regulators for VDR are presumed to be enzymes related to histone acetylation.
VDR AGONISTS AND ANTAGONISTS
VDR agonists have been generated in an effort to develop anti-osteoporotic therapies. One of the resulting weak agonists, Edirol®, has been widely applied with clinical success for the treatment of osteoporosis in Japan.[36,37] In other countries, VD is considered a dietary supplement rather than a drug, since VD has not been fully proven to be beneficial as an anti-osteoporotic therapy through extensive clinical studies.[7] The discrepancy in clinical views of VD between countries may be due in part to differences in the dietary intake of calcium.[21] As water tends to be softer (less calcemic) in Japan, it can be postulated that the anti-osteoporotic action of Edirol® is potentiated by calcium co-supplementation. Accordingly, VDR agonists like Edirol® may be more useful as anti-osteoporotic agents in areas where the water is soft and less dietary calcium is consumed.[4]
Given that VD insufficiency is closely related to the incidence of a number of diseases, the anti-inflammatory action of VD has been presumed to underlie its protective role in attenuating the onset of these diseases.[9,10] However, clinical trials have failed to prove the effectiveness of VD and its analogues in the treatment of prostate cancer, other cancers and related diseases. Since such VD analogues have been shown to significantly attenuate proliferation in cancer model cell lines, [18,20] failure of the clinical trials can be inferred to be due, at least in part, to the need to limit doses of VD analogues to avoid hypercalcemia.[21,22] Therefore, synthetic VD analogues that exert beneficial anti-inflammatory action in the absence of hypercalcemic activity are desired.
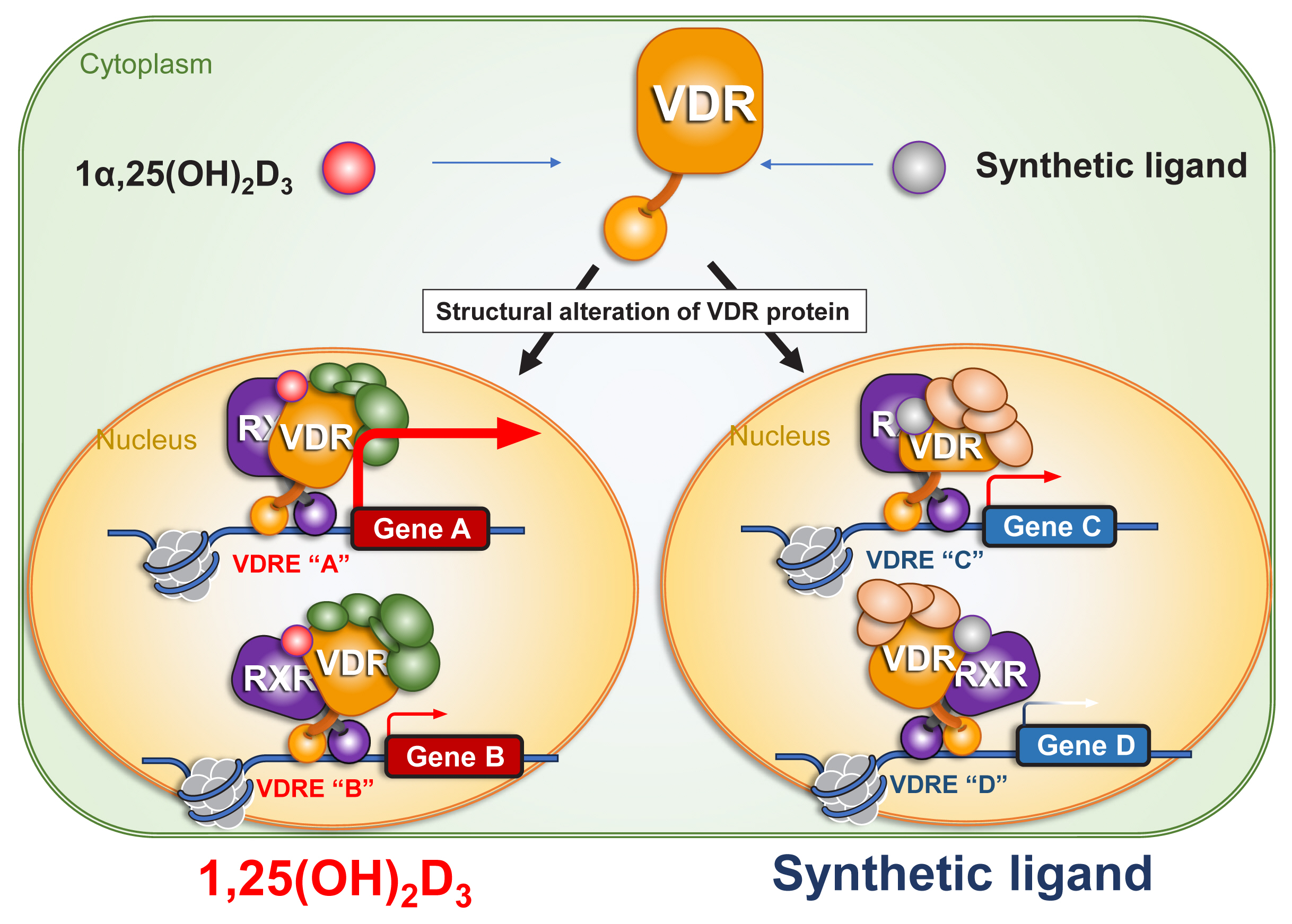
A related approach to the issue of hypercalcemia is the development of a VDR antagonist that would mitigate the hypercalcemic action of VD analogues without influencing the beneficial actions of VD for disease prevention. Recently, Rovito et al. [38] developed a VD analogue (ZK168281) to antagonize the action of VD. This compound partially trapped the VDR in the cytosol, and thereby antagonized VD action in gene regulation. The molecular basis for this activity appeared to involve differences in structural alterations to the ligand-binding domain (LBD) as induced upon binding of 1α,25(OH)2D3 and ZK168281 (Fig. 3). Interestingly, the hypercalcemic action induced by pharmacological doses of 1α,25(OH)2D3 in intact mice was blunted by this compound.[38] The authors proposed that this compound would have possible clinical applications for hypercalcemic patients, such as in cases of idiopathic infantile hypercalcemia, [25,26] but the potential for ZK168281 to attenuate inflammation remains to be studied.
BONE-SPECIFIC ACTIONS OF SYNTHETIC NR LIGANDS
The molecular mechanisms underlying the anti-inflammatory action of 1α,25(OH)2D3 remain unclear. However, the development of a partial VDR antagonist without its hypercalcemic action would be expected to realize the potential of the beneficial actions of 1α,25(OH)2D3, as administration of such a compound at higher doses would be possible. Full VDR antagonists appear detrimental to bone health, but a partial VDR antagonist could support bone health by not severely affecting calcium homeostasis.
Such an idea is supported by the clinal success of selective estrogen receptor (ER) modulators (SERMs) as anti-osteoporotic drugs.[39,40] The first generation SERM was raloxifene, and it has been administered to osteoporotic patients worldwide. Raloxifene was developed following epidemiological observations of patients with estrogen-dependent breast cancer, in which treatment with an ER-α antagonist (tamoxifen) unexpectedly did not negatively impact bone health; actually, some subjects experienced increases in bone mass. Importantly, tamoxifen had been considered at that time to be a full ER antagonist, [40] but these observations suggested that the definition of synthetic NR ligands, dependent on limited numbers of biological assays, is subject to error. Modern technologies then led to the clarification of the definition of synthetic NR ligands [41]; for example, a reporter gene assay that assessed the transactivation function of ER-α later showed that tamoxifen is an ER partial antagonist/antagonist.[42] Consistent with the findings of the molecular characterization of tamoxifen, raloxifene was also shown to be an ER partial antagonist/agonist. This drug supports calcium homeostasis in the musculoskeletal and cardiovascular systems while it exerts antagonistic activity in estrogen-dependent reproductive organs, including breast tissue and the uterus.[40]
Using similar therapeutic approaches, selective androgen receptor modulators (SARMs) have been generated, with the expectation that they would support the anabolic action of androgens in the musculoskeletal system but antagonize the action of androgens in proliferative cancer tissues.[43,44] Androgens serve as hormones that potently induce the production of muscle and bone mass, but over-production of androgen can serve as a major risk factor for androgen-dependent prostate cancer. The aberrant production of androgen can also lead to the development of male-associated characteristics in subjects who are genetically female. Thus, SARMs offer the attractive possibility of drugs that block the negative effects of androgens while supporting their positive effects. In particular, the clinical application of SARMs to older patients with sarcopenia is highly desired; such a strategy has not yet been successful in clinical trials.
Using the mechanisms of SERMs and SARMs as a model, we speculate that modern assays will be capable of identifying selective VDR ligands. Such ligands would act as agonists in the anti-inflammatory roles of VD while having a much weaker impact on the roles of VDR in calcium homeostasis. These assays thus would be capable of uncovering as-yet-unknown molecular functions of VD, and hence may constitute a foundation from which to screen for novel VDR-based therapies.
MOLECULAR BASIS OF BONE-SPECIFIC ACTIONS OF SEX STEROID HORMONE ANALOGUES
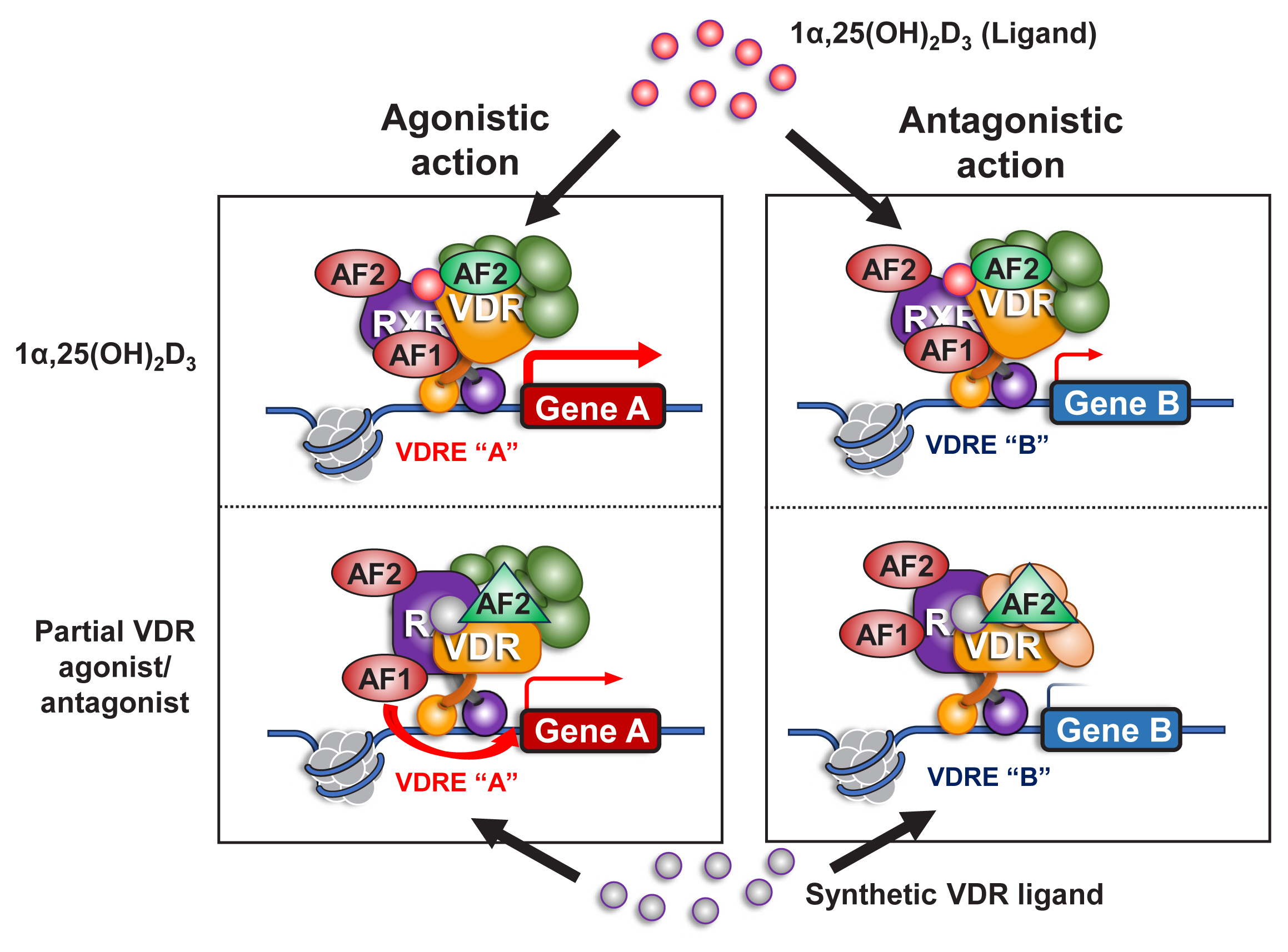
The tissue-specific actions of SERMs and SARMs have been inferred by molecular dissections of sex steroid receptors bound to synthetic ligands.[45,46] AR and the ERs form homodimers, and the monomers each possess 2 domains with ligand-dependent transactivation functions, the N-terminal activation function (AF) 1 and C-terminal AF2 domains. The AF2 domain is bifunctional, as it also serves as the LBD; it consists of 12 α-helical loops with a narrow hydrophobic pocket that specifically binds to the ligand.[35]
Upon ligand binding, α-helix 12, which is located near the C-terminal end of the protein, is drastically shifted, leading to structural alterations throughout the entire receptor. Such ligand-induced structural alterations of NR proteins lead to the dissociation of transcriptional co-repressors and the recruitment of co-activators, thus achieving ligand-induced transactivation.[33-35] As expression levels of transcriptional co-regulators are tissue- and cell-specific, ligand binding-dependent association and dissociation of transcriptional co-regulators occur in a cell-type-specific manner.[29,33] Consequently, the combinations of co-regulators that interact with the AF1 and AF2 domains of NRs in different cells reflect cell type-specific activities of these domains.
Structural analyses have shown that the binding of synthetic ligands shifts α-helix 12 of NRs to an angle distinct from that achieved upon cognate hormone binding, leading us to propose ligand type-specific conformational alterations over the entire receptor protein.[45,46] X-ray crystallographic analyses of the ER-α LBD bound to either raloxifene or estradiol provided the first such evidence,[47] and the findings correlated with the antagonistic action of raloxifene for ER-α AF2.[42] Similarly, the dominant function of AF1 over AF2 of ER-α in estrogenic action in bone tissue was demonstrated by using mouse mutants expressing only the ER-α AF1 or AF2 domain. The beneficial actions of raloxifene, presumably as well as those of the other SERMs, for bone were thus interpreted as resulting from the activation of ER-α AF1 (Table 1).
The molecular bases of the anabolic actions of androgens and SARMs largely remain elusive.[43,44] AR also has 2 functional domains, but the activity of AF2 is marginal when compared with that of AF1. Notably, the N-terminal A/B domain, which contains AF1, in AR is longer than that of ER-α.[35] This property of the ligand-induced transactivation function of AR may be an obstacle to the generation of tissue-specific SARMs.
DIFFERENTIAL GENE REGULATION BY A VD AND SYNTHETIC VDR LIGANDS
Unlike ER-α and AR, VDR contains a very short A/B domain, and this domain appears to lack a functional AF1 domain.[26,27] However, as VDR forms a heterodimer with one of the RXRs (RXRα, RXRβ, or RXRγ) to produce a ligand-dependent transactivation unit, the AF1 function of the partner RXR presumably facilitates the tissue-specific action of VD-liganded VDR.[48] It is feasible to generate a partial VDR antagonist or agonist with a minimal effect on hypercalcemia but that retains beneficial action in bone and other tissues (Fig. 4).
The desirable tissue-specific action of a VDR ligand would need to exert a different structural alteration of the VDR-containing heterodimer than that exerted by binding to endogenous 1α,25(OH)2D3. Such a possibility appears achievable in light of a recent report that showed that the structural alteration induced in the VDR LBD was different when ZK168281 or VD was bound.[38] In this regard, we recently aimed to develop a VDR antagonist, and we investigated the properties of the candidate molecule DLAM-2b. In human cell lines endogenously expressing VDR, the expected antagonistic action of DLAM-2b against VD with respect to the known VD target gene CYP24A1 was observed. By transcriptome analyses of the cultured human cells, DLAM-2b was also found to attenuate the 1α,25(OH)2D3-induced expression of a group of target genes; however, not all of the target genes known to be regulated by 1α,25(OH)2D3 were impacted. Moreover, DLAM-2b was found to alter the expression of genes that are not subject to regulation by 1α,25(OH)2D3, suggesting that VDR-bound DLAM-2b is able to interact with additional genes.
Since the altered structures of NRs bound to synthetic ligands have been shown to remodel chromatin organization, thereby leading to tissue-specific gene regulation,[35,49,50] we then used an ATAC-seq approach to compare chromatin remodeling in the presence of DLAM-2b or 1α,25(OH)2D3. However, chromatin reorganization was not observed in the cells treated with either of the 2 ligands; therefore, VDR is unlikely to be capable of chromatin remodeling. As the accessibility of the DNA at the activated gene loci was differentially regulated by the 2 ligands, histone modifications, such as acetylation and deacetylation, are more likely to facilitate gene regulation by the ligands. From these observations in human cells, we propose that DLAM-2b is a partial antagonist/antagonist for VDR, though an off-target effect not mediated by VDR on a certain set of regulated genes cannot be excluded. Moreover, further work is required to determine if DLAM-2b exerts tissue-specific actions related to 1α,25(OH)2D3-regulated events in intact animals.
CONCLUSION
VD exhibits a wide variety of biological actions, and dietary deficiency of VD underlies the incidence of multiple diseases. The molecular bases of VD action, as mediated by VDR, under normal conditions have been extensively studied, but the roles of VD and VDR in pathological settings remain largely unclear. As information regarding these systems emerges, and given the beneficial effects of VD in disease prevention, development of synthetic VD analogues with lowered hypercalcemic activity has become more promising. We expect that such compounds will be effective drugs for maintaining bone health while slowing the onset of diseases.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print