Functional Role of Phospholipase D in Bone Metabolism
Article information

Abstract
Phospholipase D (PLD) proteins are major enzymes that regulate various cellular functions, such as cell growth, cell migration, membrane trafficking, and cytoskeletal dynamics. As they are responsible for such important biological functions, PLD proteins have been considered promising therapeutic targets for various diseases, including cancer and vascular and neurological diseases. Intriguingly, emerging evidence indicates that PLD1 and PLD2, 2 major mammalian PLD isoenzymes, are the key regulators of bone remodeling; this suggests that these isozymes could be used as potential therapeutic targets for bone diseases, such as osteoporosis and rheumatoid arthritis. PLD1 or PLD2 deficiency in mice can lead to decreased bone mass and dysregulated bone homeostasis. Although both mutant mice exhibit similar skeletal phenotypes, PLD1 and PLD2 play distinct and nonredundant roles in bone cell function. This review summarizes the physiological roles of PLD1 and PLD2 in bone metabolism, focusing on recent findings of the biological functions and action mechanisms of PLD1 and PLD2 in bone cells.

INTRODUCTION
Bone is a metabolically active and highly dynamic tissue. Throughout adult life, bones undergo continuous reconstruction in a process called bone remodeling. This is a strictly controlled process coordinated by 2 main types of bone cells, osteoblasts, and osteoclasts.[1–4] Osteoblasts, derived from mesenchymal stem cells, play a crucial role in the synthesis and mineralization of bone, whereas osteoclasts, which originate from hematopoietic stem cells, are responsible for bone matrix degradation. During bone remodeling, old and brittle portions of bones are resorbed by osteoclasts and replaced with newly formed bones by osteoblasts. A fine balance between bone resorption and bone formation must be maintained to preserve bone mass and integrity and mineral homeostasis. However, derangement in bone remodeling leads to the development of osteoporosis, which is a major public health issue.
Osteoclasts are exclusive multinuclear cells derived from the monocyte/macrophage lineage progenitor cells. The proliferation, differentiation, and fusion of their precursor cells result in the formation of these polykaryons. These processes are controlled by 2 essential cytokines, macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor-κB (RANK) ligand (RANKL). RANKL induces the differentiation of osteoclasts by binding to its corresponding receptor, RANK.[5–8] This binding mediates the activation of nuclear factor-κB, p38, c-JUN N-terminal kinase, and extracellular signal-regulated kinase pathways.[9] These signaling molecules then induce and activate nuclear factor of activated T cells cytoplasmic 1 (NFATc1),[10,11] leading to osteoclast differentiation. While RANKL regulates osteoclast differentiation, M-CSF contributes to osteoclast development by promoting the proliferation, survival, and motility of osteoclast precursor cells and accelerating cytoskeletal organization.[12–14] M-CSF function is mediated by its receptor c-Fms (also known as CSF-1R) through the dimerization and autophosphorylation of c-Fms and the subsequent activation of Akt and ERK.[15]
Phospholipase D (PLD) enzyme belongs to the phospholipase superfamily that catalyzes phosphatidylcholine, the most abundant phospholipid component of cell membranes, to produce choline and phosphatidic acid.[16–18] Mammalian cells have 2 major PLD isozymes, PLD1 and PLD2.[19–21] These isozymes have a similar domain structure, with approximately 50% amino acid sequence homology, and contain several conserved domains (Fig. 1). They possess 2 conserved HKD catalytic motifs essential for enzymatic activity. Other conserved regions of PLD1 and PLD2 include the phox-homology and pleckstrin homology domains, which are known to mediate membrane interactions and control proper PLD localization. However, PLD1 differs from PLD2 because it has a unique loop region that is not present in PLD2. Although these enzymes exhibit high structural similarity, their subcellular localization is significantly different. PLD1 is generally localized in the inner membranes of cells, such as lysosome, endosome, autophagosome, and Golgi complex.[22–24] In contrast, PLD2 is primarily localized in the plasma membrane.[21,25,26] The different subcellular distribution of these isozymes may reflect their distinct biological functions.
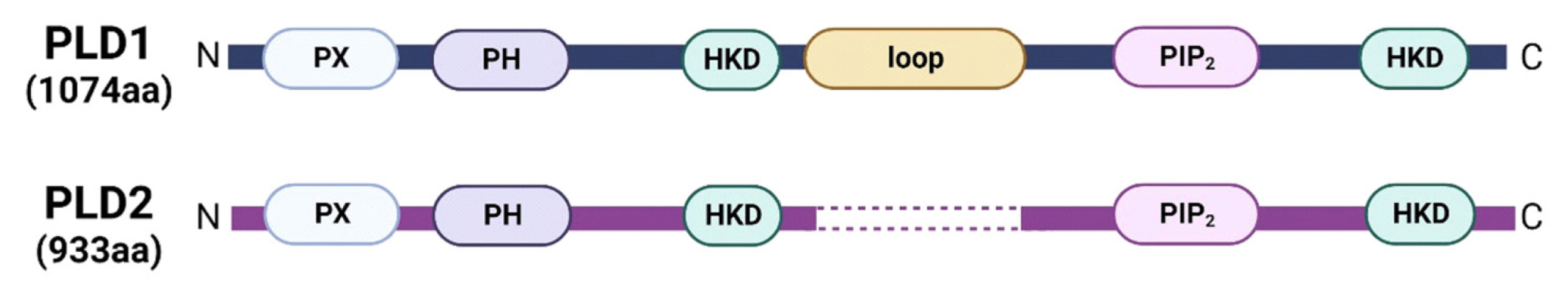
Basic structure of phospholipase D (PLD) isozymes. PLD1 and PLD2 comprises phox-homology (PX) and pleckstrin homology (PH), the catalytic HKD motif (HKD), phosphatidylinositol bisphosphate (PIP2), and PLD1 loop region.
PLD plays a crucial role in various cellular functions, such as proliferation, migration, cytoskeleton dynamics, vesicle trafficking, endocytosis, exocytosis, and receptor signaling. Disruption in these functions may lead to various disease states, such as cancer, hypertension, and neurodegenerative disease, thus making PLD a promising therapeutic target for such diseases.[16,17,27–31] Furthermore, accumulating evidence implicates PLD in the modulation of bone cell function and bone remodeling. The potential role of PLD in bone tissue has been proposed based on the results of cell-based in vitro studies. Moreover, recent studies using knockout mouse models have clarified the pivotal role of PLD1 and PLD2 in bone homeostasis, thus improving our understanding of their role in bone diseases such as osteoporosis.[32,33] This review summarizes the role of PLD1 and PLD2 in skeletal homeostasis and the diverse mechanisms through which PLD-mediated signaling contributes to different aspects of bone cell function.
ROLE OF PLD IN BONE HOMEOSTASIS
Osteoporosis is a metabolic skeletal disease caused by an imbalance between bone resorption and bone formation. Studies using PLD1- or PLD2-deficient mice have demonstrated that PLD plays a critical role in bone homeostasis. Twelve-week-old Pld1−/− mice exhibited decreased bone mass, trabecular number, and thickness.[33] A similar phenotype was observed in Pld2−/− mice, which showed a reduced bone mass at 8 and 16 weeks of age compared with their WT littermates.[32] The osteoporotic phenotype of Pld2−/− mice was further confirmed to be present in their lumbar vertebrae. These findings indicate that both PLD1 and PLD2 are important regulators of bone homeostasis and remodeling. In the next sections, we briefly outline how PLD participates in bone homeostasis through various mechanisms, including osteoclast differentiation, migration, and cytoskeletal organization as well as osteoblast differentiation.
1. Role of PLD in osteoblasts
Numerous studies have indicated that PLD is activated and regulated by various agonists, such as hormones, growth factors, and cytokines, in various cell types. The potential role of PLD in osteoblasts was initially proposed based on the results of studies on the effects of agonists on PLD activity. Prostaglandins (PGs) play an important role in bone metabolism by stimulating bone formation and resorption. [34] In osteoblast-like MC3T3-E1 cells, PLD is activated by PGD2,[35] PGE2,[36] and PGF2α.[37,38] Growth factors, such as platelet-derived growth factor [39] and basic fibroblast growth factor,[40] which are known to promote osteoblast proliferation, also activate the PLD signaling pathway in MC3T3-E1 cells. Studies on primary osteoblastic cells further showed that PLD is activated by epidermal growth factor (EGF) [41] and that EGF-induced proliferation of these cells requires PLD activation.[42] Parathyroid hormone, an anabolic agent and a key regulator of bone remodeling, also stimulates PLD activation in UMR-106 osteoblastic cells. [43,44] Furthermore, MG63 osteoblast-like cells cultured on titanium surfaces exhibited increased PLD activity, alkaline phosphatase (ALP) activity, and osteocalcin production. [45] Together, these findings indicate the potential role of PLD in osteoblast differentiation.
As mentioned above, most previous studies focused on the effect of various agonists on PLD activation. However, recent studies have demonstrated the expression pattern of PLDs and their functional role in osteoblasts. The mRNA and protein expression levels of PLD1 were upregulated during the differentiation of murine primary osteoblasts [46] and human mesenchymal stem cells (MSCs).[33] In contrast, PLD2 expression was low [33] and did not change significantly during the osteogenic process.[33,46] PLD1 overexpression in Saos-2 cells stimulated the mineralization process, whereas PLD2 overexpression did not affect calcium nodule formation.[46] Conversely, PLD1 knockdown in human MSCs decreased mineral deposition, ALP activity, and osteogenic marker expression.[33] Pharmacological suppression of PLD1 through its selective inhibitor reduced osteogenic differentiation.[46] However, treatment with a PLD2-selective inhibitor had no significant effect.[46]
The physiological function of PLD in osteoblasts was clearly demonstrated by studies on PLD-deficient mice. MSCs derived from Pld1−/− mice exhibit impaired osteogenic potential and reduced expression of osteogenic markers, including Runx2 and osteocalcin.[33] In contrast, Pld2−/− mice did not show any differences in osteoblast parameters, such as bone formation rate and mineral apposition rate, compared with WT mice, demonstrating that PLD2 is not implicated in the osteoblast bone-forming process.[32] Taken together, these findings indicate that PLD1, but not PLD2, plays a crucial role in bone mineralization in vivo as well as in vitro.
2. Role of PLD in osteoclasts
Early studies on PLD signaling in osteoclasts indicated its potential role in pathological conditions, such as cancer metastasis and rheumatoid arthritis (RA). Bone is a common metastasis site for lung, breast, and prostate cancers, which can lead to severe morbidity. Cancer cells produce numerous substances that induce their migration to the bone and stimulate osteoclastic bone resorption. For example, the expression of an inflammatory cytokine interleukin-8 (IL-8) is elevated in human lung adenocarcinoma and breast cancer cells, thus promoting osteolysis.[47] Treatment of peripheral blood mononuclear cells (PBMCs) with recombinant human IL-8 or their exposure to conditioned media derived from human lung cancer cells increases osteoclast formation and function. In addition, PBMCs treated with the serum of patients with lung cancer induce osteoclast formation. This increase in osteoclast formation was associated with elevated PLD activity. Blocking PLD using its specific inhibitor reduces osteoclast formation.[48] These findings suggest that PLD is involved in osteoclastic bone resorption mediated by lung cancer-derived IL-8.
Although PLD has been suggested to stimulate osteoclast formation under pathological conditions, recent studies on mice models of Pld1−/− and Pld2−/− have provided further insight into the direct physiological role of PLD in osteoclasts.[32,33]
1) PLD and osteoclast differentiation
Pld1-deficient mice exhibit reduced bone mass because of increased osteoclast differentiation as well as impaired osteoblastogenesis.[33] Pld1−/− preosteoblasts (pre-OBs) cocultured with WT bone marrow macrophages (BMMs) or WT pre-OBs cocultured with Pld1−/− BMMs leads to an increase in the number of tartrate-resistant acid phosphatase (TRAP)-positive osteoclasts and upregulates the expression of osteoclastogenic markers relative to that of WT pre-OBs cocultured with WT BMMs. Pld1−/− pre-OBs cocultured with Pld1−/− BMMs results in the highest increase in osteoclast number compared with other groups. Consistently, in vivo TRAP staining revealed that osteoclast number increased in Pld1−/− mice compared with WT mice. Mechanistically, accelerated osteoclast differentiation in PLD1 deficiency condition is associated with increased RANKL/osteoprotegerin (OPG) ratio in osteoblasts and the upregulation of peroxisome proliferator-activated receptor-γ (PPARγ) and CCAAT/enhancer binding protein-α (C/EBPα)-induced c-Fos expression in osteoclasts. Osteoblasts produce RANKL and OPG, a decoy receptor for RANKL, both of which affect osteoclastogenesis. Pld1−/− osteoblasts exhibit an increased RANKL/OPG ratio, enhancing osteoclast differentiation. PPARγ [49] and C/EBPα [50] function as direct upstream regulators of c-Fos, an essential modulator of osteoclastogenesis. Osteoclast-specific deletion of PPARγ using a Tie2-Cre/flox mouse model indicated increased bone mass in mice due to defective osteoclastogenesis.[49] Similarly, C/EBPα-deficient newborn mice develop osteopetrosis owing to impaired osteoclast differentiation.[50] PLD1 deficiency not only increases the expression of PPARγ and C/EBPα but also increases the binding capacity of PPARγ and C/EBPα to the c-Fos promoter as well as its promoter activity. Together, these findings indicate that accelerated osteoclast differentiation in PLD1 deficiency condition is due to the combined action of osteoclasts and osteoblasts. Further, PLD2 deficiency does not affect osteoclast differentiation, as evidenced by the unaltered expression of osteoclastogenic markers including c-Fos, NFATc1, and TRAP, as well as unaltered RANKL signaling.[32]
2) PLD and osteoclast migration
Cell migration is essential for several biological functions and is a highly dynamic event that involves remodeling of the actin cytoskeleton. In particular, the migration of osteoclastic cells is crucial for osteoclast-mediated bone resorption. It is well recognized that PLD acts as a major player in the migration of various cell types, such as cancer and inflammatory cells. In osteoclasts, PLD2 participates in cell migration.[32] PLD2 deficiency promotes M-CSF–induced migration of osteoclast lineage cells, including BMMs and preosteoclasts.
Phosphoinositide 3-kinase (PI3K) is a key signaling molecule involved in cell migration of various cells and is required for Akt activation. For example, dual Ig domain-containing adhesion molecule suppresses endothelial cell migration by inhibiting the PI3K/Akt pathway.[51] Deletion of the p85α subunit of class I PI3K results in impaired osteoclast migration toward αvβ3 integrins or osteopontin in the presence of M-CSF.[52] Consequently, p85α-deficient mice have reduced bone resorption ability, increasing bone mass.[52] Exposure of BMMs to a PI3K specific inhibitor LY294002 decreases the ability of M-CSF to stimulate BMM migration.[32] These findings indicate that PI3K/Akt signaling is critical for the migration of osteoclastic cells. PLD2-deficient BMMs exhibit increased activation of Akt signaling in response to M-CSF.[32] Thus, PLD2 negatively regulates osteoclastic cell migration by modulating M-CSF-induced PI3K/Akt signaling pathway. The function of PLD1 in osteoclastic cell migration has not been investigated yet.
3) PLD and osteoclast cytoskeleton
Degradation of the bone matrix is a hallmark action of osteoclasts. During bone resorption, osteoclasts attach tightly to the bone surface and reorganize their cytoskeleton to generate F-actin ring or sealing zone.[53,54] These unique and specialized structures are required for the bone-resorptive function of mature osteoclasts. To degrade the bone matrix, osteoclasts secrete protons and enzymes, such as cathepsin K and matrix metalloproteinases, through the ruffled border, which is surrounded by actin rings. The size of osteoclasts and actin ring indicates the capacity of osteoclasts to resorb the bone matrix. PLD2 deficiency increases osteoclast size and leads to larger actin ring formation in vitro.[32] Consistently, in vivo TRAP staining revealed an increased osteoclast surface per bone surface in Pld2−/− mice. The formation of actin rings in osteoclasts depends on an intact microtubule network, specifically on the acetylated form of microtubules.[53–55] Reflecting their enlarged actin rings, Pld2−/− osteoclasts have more acetylated microtubules than WT osteoclasts.[32]
PI3K/Akt pathway is central for actin remodeling as well as cell migration. Osteoclasts derived from mice lacking the p85α subunit of PI3K failed to form actin rings from actin clusters.[52] Osteoclast-specific deletion of p85α and β subunits results in high bone mass due to defective ruffled border formation and vesicle transport.[56] Targeted deletion of Akt1 and Akt2 in osteoclasts causes failure of actin ring formation, resulting in the formation of an osteosclerosis phenotype.[57] Treatment with Akt inhibitor in osteoclasts reduces the expression level of acetylated tubulin.[57] Akt promotes osteoclast actin remodeling by inhibiting glycogen synthase kinase-3β (GSK-3β) phosphorylation. GSK-3β then regulates microtubule stability by modulating the interaction of microtubules with microtubule-associated proteins, such as adenomatous polyposis coli, end-binding protein 1, and dynactin.[57]
Overall, these studies indicate that PI3K/Akt/GSK-3β signaling controls osteoclast bone-resorptive function by modulating actin ring formation and microtubule stability. PLD2 deficiency enhances Akt phosphorylation induced by M-CSF, but not RANKL.[32] In Pld2−/− osteoclasts, GSK-3β was hyperphosphorylated. Furthermore, increased microtubule acetylation in Pld2−/− osteoclasts was restored after treatment with Akt inhibitor. Mechanistically, PLD2 binds with the M-CSF receptor (c-Fms) and PI3K and blocks M-CSF–mediated PI3K/Akt/GSK-3β pathway, which in turn inhibits the migration of osteoclast lineage cells and cytoskeletal organization in mature osteoclasts.[32] The role of PLD1 in osteoclastic cell migration and cytoskeletal organization has not been investigated yet.
CONCLUSION
In this review, we discussed the role of the PLD family proteins PLD1 and PLD2 in bone metabolism and found that they are key modulators of bone homeostasis. Deficiency of either PLD1 or PLD2 results in a similar osteoporotic phenotype. However, these isozymes play distinct roles in bone formation and bone resorption. PLD1 positively regulates osteoblast differentiation but negatively modulates osteoclastogenesis (Fig. 2). In contrast, PLD2 is not essential for bone formation but is involved in osteoclast function. PLD2 acts as a negative regulator of osteoclastic bone resorption by modulating cell migration and microtubule stability (Fig. 3). The function of PLD is known to be mediated through its enzymatic activity or direct interaction with other signaling molecules. Thus, PLD can control bone cell function, presumably in an activity-dependent manner or by acting as an adaptor molecule. Therefore, given the pivotal role of the PLD family in bone metabolism, the design of novel compounds capable of modulating the enzymatic activity or the interaction between PLD and signaling proteins will provide promising therapeutic potential for various bone diseases, such as osteoporosis, RA, and osteoarthritis.

Schematic diagram of phospholipase D1 (PLD1) action in bone cells. PLD1 induces osteoblast differentiation by upregulating Runt-related transcription factor 2 (Runx2) and suppresses osteoclast differentiation by downregulating receptor activator of nuclear factor-κB ligand (RANKL)/osteoprotegerin (OPG) ratio and peroxisome proliferator-activated receptor-γ (PPARγ) and CCAAT/enhancer binding protein-α (C/EBPα)-mediated c-Fos expression. NFATc1, nuclear factor of activated T cells cytoplasmic 1.

Schematic diagram of phospholipase D2 (PLD2) action in bone cells. In osteoclasts, PLD2 forms a complex with the macrophage colony-stimulating factor (M-CSF) receptor (c-Fms) and phosphoinositide 3-kinase (PI3K) and antagonizes cell migration and microtubule stability by attenuating the Akt/glycogen synthase kinase-3β (GSK-3β) signaling pathway, thereby leading to impaired bone resorption. On the other hand, PLD2 does not affect bone formation.
Notes
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean Government (MSIT) (NRF-2022R1A2C1006105, NRF-2018R1A2B 6001298) and Basic Science Research Program through the NRF funded by the Ministry of Education (NRF-2021R1I1A1A01051983).
Ethics approval and consent to participate
Not applicable.
Conflict of interest
No potential conflict of interest relevant to this article was reported.