INTRODUCTION
Vitamin D (VD) is a pivotal hormone/vitamin controlling calcium homeostasis, but also regulating a number of biological processes.[1] VD exerts a wide variety of biological actions through nuclear VD receptor (VDR)-mediated transcriptional regulations of the target genes, and the target gene products facilitate the biological effects.[2,3,4] Clinically, VD as well as the related compounds is successful as an anti-osteoporotic agent, drawing huge attention to the molecular basis of the VD actions in the bone field.[5] Ligand-dependent transcriptional by VDR is coupled with chromatin reconfiguration/epigenetic alteration.[3,4] Recent progress of whole human genome analysis has uncovered the presence of numerous of non-coding RNAs (ncRNAs) and tertiary structure of chromatin and their significance in gene regulation.[6] As the certain group of the non-coding RNA genes are transcribed by RNA polymerase II,[7] such ncRNAs could be VD/VDR target genes. In this short review, the role of enhancer RNA (eRNA), a newly emerged class of ncRNAs, in the gene regulations is reviewed, and possible contribution of eRNA to the VDR-mediated epigenetic regulation is discussed.
BIOLOGICAL ROLE OF THE NUCLEAR VDR)
1. The VDR mediates the biological actions of VD
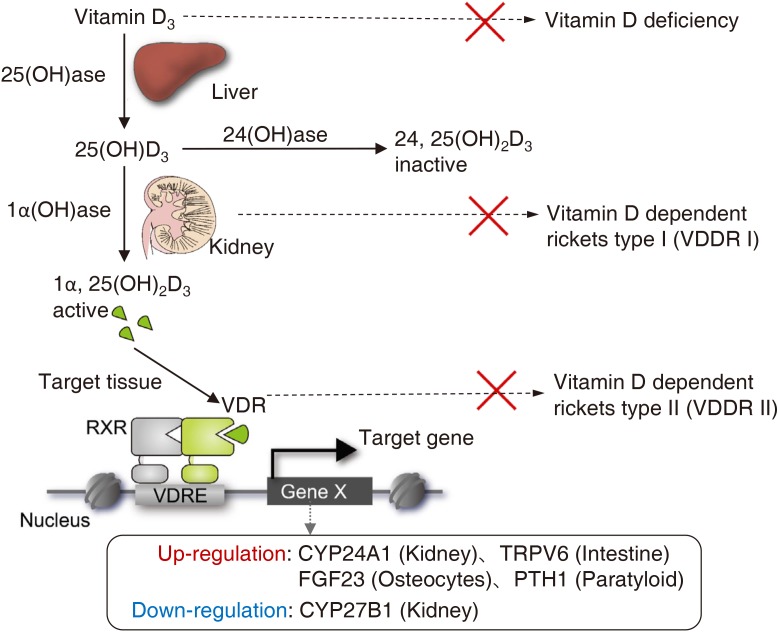
VD is a fat-soluble vitamin normally present as a nutrient in balanced diets; it can also be biosynthesized in the body in response to ultraviolet light exposure. In vertebrates, VD exerts a wide variety of biological actions and plays a pivotal role in calcium homeostasis.[1] Owing to its significance in physiological processes, an excess or a deficiency of VD leads to severe pathological conditions, including diseases like rickets.[2] The biologically active form of VD, 1α,25-dihydroxy-vitamin D3 (1α,25[OH]2D3), is produced through 2 hydroxylation steps in the liver and then the kidney. Hepatic CYP24B1 hydroxylates the precursor of VD into 25(OH)D3 followed by its conversion into the active VD form through the second (1α-hydroxylation) by the renal enzyme CYP27B1.[1,8] Loss-of-function of CYP27B1 by genetic mutations causes type I hereditary rickets, however these patients can be rescued by administration of precursor 1α(OH)D3 or 1α,25(OH)2D3.[9] Excess VD activates kidney CYP24A1, resulting in the conversion of 1α,25(OH)2D3 into 24,25(OH)2D3, a biologically inactive form of VD (Fig. 1).[1,8]
Most of the known biological actions of VD are mediated by the nuclear VDR.[3,4,10] The VDR belongs to the nuclear receptor (NR) gene superfamily comprised of 48 currently known members.[11] Similar to other family members, the VDR acts as a ligand-dependent DNA-binding transcriptional factor. For DNA binding, the VDR requires cooperation with one of the 3 subtypes (α, β, or γ) of another NR superfamily member, the retinoid X receptor (RXR) and formation of VDR-RXR heterodimers.[3,4] A consensus DNA binding site for VDR-RXR is composed of 2 AGGTCA direct repeats (DR) spaced by 3 nucleotides (a so-called DR3-type motif sequence), 5′-AGGTCANNNAGGTCA-3′. This DNA sequence serves as an efficient VD-responsive transcriptional enhancer named the VD response element (VDRE).[3,4]
ChIP-sequencing (ChIP-seq) analyses have shown that the numbers and locations of genomic VDR binding sites differ significantly between different cell types. Surprisingly, only a minority of identified genomic VDR binding sites contain functional variants of the VDRE consensus sequence, [12] suggesting that VDRE-independent chromatin binding of VDR might be mediated by various cell-specific regulatory factors. It has been shown that for chromatin binding, sex steroid receptors (androgen or estrogen receptor homodimers) require an association with so-called pioneering factors, such as FOXA1.[13,14] Therefore, it is likely that similar, yet to be identified regulatory factors modulate (i.e., facilitate or inhibit) VDR binding to chromatin regardless of genomic VDRE motif sequences. In this regard, as a candidate, co-localization of PU.1 with VDR on chromatin was reported.[15]
2. VDR is a VD-dependent transcription factor
Although the VDR may associate with chromatin and specific DNA elements even in the absence of VD, the ligand is required for the VDR to regulate target gene transcription (Fig. 2).[3] VD-induced transcription requires the assembly of multi-subunit protein complexes at the target gene promoters.[16] These complexes include the transcription initiation complex (RNA polymerase II plus basal transcription factors) and a mediator complex (MED) bridging the promoter-bound VDR with the transcription initiation complex.[17,18]
In the absence of ligand, the VDR in the nucleus appears to associate with transcriptional co-repressors. For VD-induced modulation of gene expression, functionally opposing VDR transcriptional co-regulators sequentially dissociate and associate upon ligand biding. In transactivation, co-repressors that are associated with the VDR dissociate and are replaced by co-activator complexes. In VD-dependent transrepression, dissociation of co-repressors appears to be aborted and/or different types of co-repressors are recruited by the VDR in a ligand-dependent manner.[19,20] However, in comparison with transactivation, there is uncertainty regarding the molecular events that occur during ligand-induced transrepression.
3. The VDR is essential for bone growth and health
Lack-of-function VDR mutations lead to development of severe skeletal abnormalities known as hereditary type II rickets.[2,10] In contrast to type I rickets,[9] neither 1α,25(OH)2D3 nor 1α(OH)D3 can rescue severe rachitic phenotypes in type II rickets patients.[2,10]
Type II rickets phenotypes can be experimentally recapitulated in mice by experimental VDR gene knock-out (KO), and animals with genetic VDR deficiency display severe skeletal abnormalities.[10] However, rachitic phenotypes in VDR KO mice develop after weaning.[10] This suggests that VD-VDR signaling is dispensable in nursing animals, but becomes essential after the transition to a normal diet. Remarkably, severe skeletal defects in the adult VDR KO mice can be ameliorated simply by feeding with a high calcium diet.[21] Thus, high calcium intake from nursing milk appears to protect the mutant neonates from rachitic disorders. This suggests that the VDR-mediated actions of VD in bone growth and metabolism do not target bone tissue directly, but rather mediate serum calcium concentrations sufficient for bone formation.[22,23] Indeed, an elegant study on mice with selectively ablated VDR in the intestine has shown that calcium mobilization from bone is enhanced when a hypocalcemic state was induced.[24,25]
4. Synthetic VD compounds are beneficial for bone health in hypervitaminosis
Osteomalacia is attributed to insufficient calcium intake, similar to VD deficiency,[1] as postulated from the results obtained with VDR mutant mice.[10,21,22,23,24,25] VD dietary supplementation is widely recommended to prevent a hypocalcemic state, and in the US, calcium is widely distributed through milk. Due to low levels of calcium in water in Japan and in other countries, calcium intake from the diet tends to be insufficient, particularly for the elderly who lose the ability to absorb calcium. This could explain why VD is clinically effective for bone mass increases for osteoporotic patients in Japan. Use of eldecalcitol (ELD) has been a clinically successful approach to preventing bone fracture and increasing bone mass. However, it has less of a calcemic effect than does calcitriol.[5,26] Furthermore, supplementation of ELD appears beneficial in any anti-osteoporotic drug treatment in terms of target tissues and skeletal cell types.[5,23] The other anti-osteoporotic drugs on the market modulate bone metabolism by attenuating bone resorption and/or stimulating bone formation for bone fracture prevention. In contrast, the major target tissue for ELD appears to be the intestine and kidney.[23,26] This difference in the target tissues/cells for ELD and the other drugs warrants the use of ELD as a co-treatment with other anti-osteoporotic drugs.
MOLECULAR MECHANISMS OF TRANSCRIPTIONAL AND EPIGENETIC CONTROL BY THE VDR
1. Pleiotropic tissue-specific action of VDR
The impaired growth and maintenance of bone in type II rickets patients and VDR KO mutant mice can be corrected by high levels of calcium supplementation.[14,17] However, other notable phenotypic abnormalities such as alopecia cannot be reversed by any currently known treatment. Since type I rickets patients do not exhibit hair loss in early childhood, infant alopecia is a clinical indication distinguishing type II hereditary rickets from type I.[4,17] None of the available natural or synthetic VDR agonists has been able to prevent hair loss in these patients or mutant mice.[10] However, targeted expression of a VDR transgene in the skin of VDR KO mice rescued the alopecia phenotype,[27] suggesting that epithelial follicular cells are direct target of the VDR. These and similar observations indicate that the VDR targets tissue-specific sets of genes (Fig. 1).[3,4]
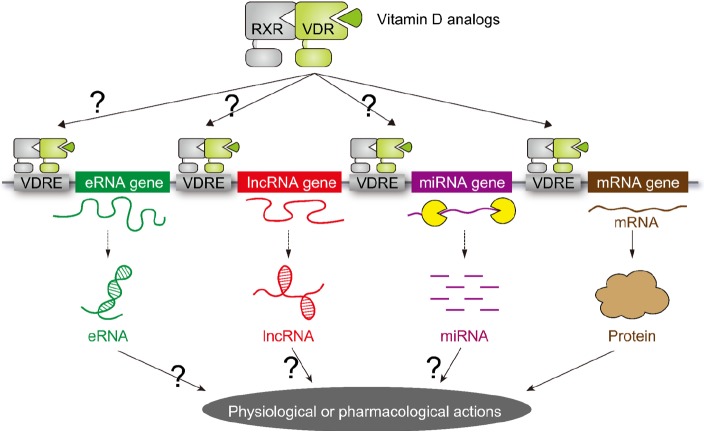
Initially, the VD-induced transcription of protein-encoding genes was assumed to account for all pleiotropic actions of VDR. However, recent mammalian transcriptome studies demonstrated that a significant fraction of the RNA polymerase II transcripts represent non-protein coding RNA species,[6,28] and ChIP-seq experiments showed that the VDR was also bound to genomic sites remote from protein encoding genes.[12] These results suggest that non-coding RNA genes might potentially be regulated by VD-VDR signaling (Fig. 2), though this idea still awaits experimental confirmation.
2. VDR-mediated transactivation involves chromatin reorganization at target loci
The VDR regulates cell type-specific sets of genes that reflect differences in the state of chromatin at the target loci between cells of different tissues. Arrays of target genes also change during cell differentiation. Such spatial and temporal expression of genes targeted by the VDR is achieved through highly regulated chromatin remodeling and histone modification.[29,30] The ordered alterations of chromatin are controlled by a range of nuclear protein factors that support fundamental steps of gene regulation common for all kinds of DNA-binding transcriptional factors, including the VDR.[29,30]
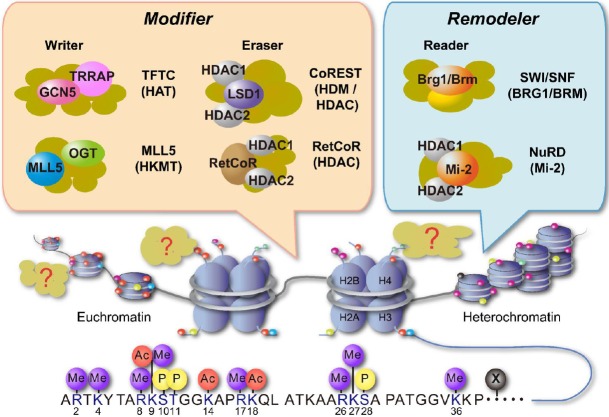
Regulated chromatin remodeling [31,32] is directed by post-translational modification (PTM) of histone proteins (Fig. 3) known as the “histone code”.[33,34,35] Histone PTMs can be reversed through the action of functionally opposing enzymes (histone modifiers). Their activities on chromatin are finely tuned by intracellular and extracellular stimuli that direct the transitions between active and repressed states of chromatin at specific genomic loci.[33,34]
Initially, histone acetylation and deacetylation were thought to direct the transition between active and repressed chromatin states.[33,34] However, in signaling cascades, several histone PTMs were identified upstream that impacted chromatin conformation.[33,35,36] Currently, specific patterns of histone methylations are regarded as pivotal determinants for downstream histone PTMs.[33,34,37] A lysine (K) residue NH3+ group can be mono-, di- or tri-methylated, and the arginine (R) NH2+side chain group can be mono- or dimethylated. Patterns of methylation at particular histone H3 lysine residues appear to be crucial in defining chromatin's state. Methylations at H3K4 and H3K36 represent reliable markers of activated chromatin, whereas methylations at H3K9 and H3K27 are markers of repressed genomic regions. The degree of methylation, or the number of methyl moieties on specific H3 lysine residues appears to vary between different functional genomic regions. Di- and tri-methylated H3K4 (H3K4me2/3) are closely associated with activated chromatin at mRNA-encoding gene loci, including their promoters.[33,34,37] Whereas H3K27m2/3 are more prevalent at silent enhancer regions of chromatin, acetylated H3K27 is associated with activated enhancer genomic regions. Apparently, repressing lysine methylation residues, like H3K27 or H3K9, can be either acetylated or methylated, thereby providing a mechanism of reversing opposing states of chromatin.[33,36] Interestingly, acetylation of activating methylation residues has not been detected and would most likely be functionally redundant. Thus, the effects of histone lysine methylation on chromatin organization are more complex than that of acetylation. Thus, histone acetylation and deacetylation invariably produce opposing results. Moreover, chromatin activation can be accomplished by both methylation (at H3K4 and H3K36) and demethylation (methylated H3K9 and H3K27); and in reverse, depending on the substrate, histone methylation (H3K9 and H3K27) and demethylation (methylated H3K4 and H3K36) would likely result in transcriptional repression. [33,34,37]
3. Histone modifiers and chromatin remodelers are transcriptional co-regulators of the VDR
The VDR exerts its regulatory effects on chromatin in cooperation
with various histone modifiers and ATPase-dependent
chromatin remodelers that function as transcriptional
co-regulators of the VDR (Fig. 3).[29,30] Generally, histone
modifying and chromatin remodeling enzymes do not act alone, but function as catalytic subunits in large multi-subunit nuclear protein complexes often exceeding 2 MDa in size (Fig. 3),[17,18,38] by aid of histone chaperones.[39,40,41]
Histone acetyltransferases (HATs) and histone deacetylases (HDACs) were the first cofactors shown to co-activate and co-repress the VDR and other NRs through ε-N lysine acetylation and ε-N-acetyl lysine deacetylation, respectively.[3,12,17,18,19,29] During VDR transactivation, HDAC complexes dissociate from ligand-receptor complexes followed by VDR recruitment of HAT activities. This basic mechanism accounts only for the relatively fast activation by NRs of euchromatic loci that were being slowly transcribed. However, the dynamics of induction of VDR target genes appear to correlate with the degree of their chromatin repression. The transactivation of repressed target loci requires more extensive multistep rewriting of histone code markers (i.e., erasing of repression and introduction of activation PTMs) leading to more profound changes in chromatin configuration. As an upstream signal, histone methylation is believed to direct the rewriting of histone PTMs through coordinated action of various histone methyltransferase (HMT) and histone demethylase (HDM) activities.[33,36,37] Guided by de novo introduced histone PTMs, recruited chromatin remodeling factors reconfigure chromatin into conformations permissive for transcriptional activation.[33,34,36] In vitro assays showed that some, but not all HMTs and HDMs directly interact with the VDR and other NRs.[20,36,42] In contrast, chromatin remodeling ATPases interact with NRs indirectly, through bridging proteins of multi-subunit remodeler complexes.[17,18]
A POSSIBLE ROLE OF ERNA IN CHROMOSOMAL RECONFIGURATION BY THE VDR
1. Dynamic chromatin reorganization supports gene regulation
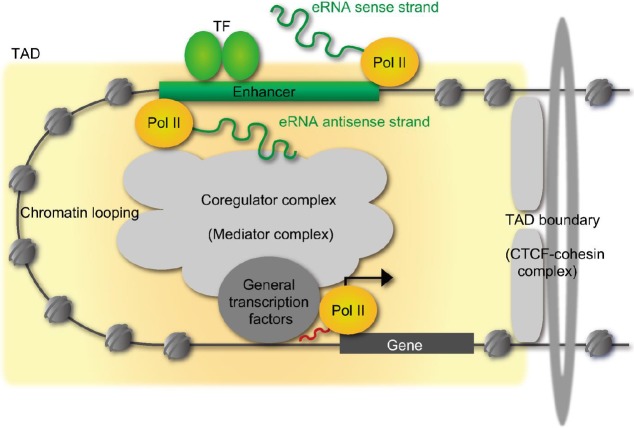
Efforts to understand cis- and trans-regulatory factors and their impact on gene activities are being extended to higher orders of chromosomal organization. Studies of interactions between genomic segments by various chromosome conformation capture techniques (Hi-C, ChIP-loop, chromatin interaction analysis with paired-end tag sequencing and related methods) have revealed that individual chromosomes are organized into topologically associating domains (TADs) made distinctive by increased intra-domain chromatin contacts.[43,44] Structurally, each TAD is flanked by TAD boundary elements and contains multiple subdomains of DNA loops. The molecular basis of TAD formation and subdomain organization and dynamics remains largely unclear. However, it has been established that the TAD DNA loops are formed and stabilized by interactions between cohesins, ring-like protein complexes better known for holding sister chromatid together, and CCCTC-binding factor (CTCF), a DNA binding protein initially identified as a transcriptional repressor.[6,44,45] The observed correlation between the distribution of specific epigenetic markers or bound RNA polymerase II and boundaries of individual TADs along the chromosome suggests that individual TADs may have different functionalities (active, repressed or inert), and that genes within the same TAD are transcribed together. The CTCF binding was indeed observed in the promoters of the VD target genes.[46] Furthermore, genome-wide studies of long-range chromatin interactions indicate that chromatin interactions are restricted within TADs, and that DNA loop subdomains provide spatial interaction between distal enhancers and promoters of regulated gene (Fig. 4).[6,45] 2. A possible role
2. A possible role of eRNA in chromatin looping and enhancer-mediated gene activation by the VDR
TADs contribute to gene regulation by modulating the interaction between DNA regulatory elements, such as enhancers, and their target genes through intra-domain DNA looping dynamics. Various nuclear proteins and their complexes have been implicated in bridging distal DNA enhancers with proximal promoters of regulated target genes. Recently, eRNA (Fig. 5) has been identified as a factor that facilitates DNA looping and intra-loop sequence interaction (Fig. 4).[6,7,45,47] Interestingly, as a trans-mechanism of regulation, eRNA has been shown to recruit cohesin to targeted gene promoters located on different chromosomes. eRNAs are transcribed by RNA polymerase II from clustered, or super enhancers (Fig. 4).[45,47] Ligand-dependent induction of eRNAs and their involvement in gene regulations were shown in the target mRNA genes for estrogen receptor α (ERα).[48,49] It is therefore, conceivable that eRNA genes may target genes for the VDR, similar to other classes of ncRNAs shown to be transcribed by RNA polymerase II, like miRNAs (Fig. 6), although no report is yet available about eRNAs' involvement in gene regulations by the VDR.
Our group is currently testing the above hypothesis using human 24-hydroxylase (CYP24A1) as a model gene, the transcription of which can be activated by VD.[4,8] In human keratinocyte HaCaT cells, we observed strong induction of endogenous CYP24A1 expression after treatment with VD.[50] Public database records show that the intergenic region downstream from the structural CYP24A1 gene harbors a super enhancer. The presence of several functional VDREs has been reported in this super enhancer region.[51]
In a preliminary screening of transcripts, we identified several eRNA candidates transcribed from the 3′-intergenic enhancer region. It has been reported that eRNAs are expressed at low levels and their half-lives are quite short due to instability.[47] Furthermore, distinct from the single start site used in mRNA transcription, eRNA genes are transcribed from multiple start sites and from both DNA strands. In our experiments, the expression levels of eRNAs transcribed from the intergenic enhancer downstream from CYP24A1 were very low. We also confirmed the presence of multiple start sites of these eRNA genes. Knock-down of one of the candidate transcripts by siRNA resulted in a significant decrease in CYP24A1 gene induction by VD in HaCaT cells. Consistent with earlier reports,[29,46,51] chromosome interaction analysis has shown a ligand-induced reconfiguration in chromatin looping of this downstream intergenic super enhancer region. Though the studied eRNAs appear to facilitate chromatin looping,[47,52] their properties and involvement in chromatin reconfiguration remain to be determined. Taken together, our findings (Nishimura et al., unpublished data) support the hypothesis that eRNAs transcribed from the intergenic super enhancer downstream from the CYP24A1 locus facilitate the induction of genes by VD (Fig. 6).
CONCLUSIONS AND PERSPECTIVES
Involvement of enhancers is a common feature in mechanisms of transactivation, including by NRs. It now appears that enhancers produce eRNA transcripts that contribute to the cis-action of DNA enhancers through trans-action regulatory mechanisms.[6,7,47] Furthermore, as RNA Polymerase II products, the expression of at least some eRNA genes may be regulated by extracellular signals, including hormones. In this review we introduced our preliminary data that suggest that eRNAs cooperate with the VDR in the transactivation of target genes, probably through induction of chromatin looping favorable for target promoter-enhancer interaction, as the chromatin looping was already seen in the promoters of the VDR target genes.[29,30,46,51] However, the exact mechanisms of eRNA action remain to be identified. Some key questions to be resolved include the following: (1) How do eRNAs induce reconfiguration of higher-order chromosomal structure? (2) What protein factors cooperate with eRNA in gene regulation? (3) Can eRNAs affect not only intra-TAD chromatin loop interaction, but also bridge inter-TAD subdomains? In this respect, the functional interaction of such eRNAs with T cell transcription factor [46] and Yin Yang 1 (YY1) [52] is interesting to be assessed in details.
Recent reports indicate that recruitment of cohesin may represent a common mechanism in the induction of chromosomal DNA looping.[6,45] Cohesin recruitment has been shown for a muscle-specific eRNA in trans-regulation of the myogenin gene,[52] and for the ubiquitous transcription factor YY1 protein in loop formation to facilitate enhancer- promoter interaction.[53] Therefore, regarding the eRNAs that we propose to be involved in CYP24A1 gene transactivation by the VDR, it would be interesting to investigate whether these eRNAs interact with cohesin to facilitate the looping and whether these eRNAs interact with VDR co-factors. This notion is based on the previous reports,[48,49] in which estrogen-induced eRNAs facilitate transcriptional regulation of the ERα target mRNA genes. Pleiotropic actions of the VDR may also depend on cooperation with cell-type specific eRNAs. Clearly, VDR target gene regulation involves novel epigenomic mechanisms at the level of chromatin conformation and TAD subdomain organization. This mechanism is expected to account for at least in part, the complicated but highly regulated process of bone cell proliferation and differentiation.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print