Origin of Osteoclasts: Osteoclast Precursor Cells
Article information

Abstract
Osteoclasts are multinucleated bone-resorbing cells and a key player in bone remodeling for health and disease. Since the discovery of osteoclasts in 1873, the structure and function of osteoclasts and the molecular and cellular mechanisms of osteoclastogenesis have been extensively studied. Moreover, it has been well established that osteoclasts are differentiated in vitro from myeloid cells such as bone marrow macrophages or monocytes. The concept showing that osteoclasts are derived from a specific population (named osteoclast precursor cells [OCPs]) among myeloid cells has been long hypothesized. However, the specific precursor population of osteoclasts is not clearly defined yet. A growing body of work provides evidence of the developmental origin and lifespan of murine osteoclasts, particularly in vivo. Here, we review the emerging evidence that supports the existence of OCPs and discuss current insights into their identity.
INTRODUCTION
Osteoclasts are multinucleated bone-resorbing cells that play an integral role in physiological bone remodeling, repair in bone injury, and pathological bone resorption commonly associated with osteoporosis or inflammatory conditions such as rheumatoid arthritis (RA) and osteolysis.[1–4] The hematopoietic origin of osteoclasts has been well established as demonstrated in several osteopetrotic mouse models.[5–7] Osteopetrosis which is mediated by defective osteoclasts in microphtalmic (mi/mi) mice is rescued by the transplantation of bone marrow (BM) or spleen mononucleated cells from normal mice, and functional osteoclasts are observed within 2 to 3 weeks after transplantation.[6] T-cell immune regulator 1 mutation in osteosclerotic (oc/oc) mice results in functionally impaired osteoclasts and increased bone mass, reversed by the transplantation of hematopoietic stem cells (HSCs).[7] In addition, it has been shown that osteoclasts can be derived in vitro from cells of a myeloid origin, including human and mouse BM cells, splenic macrophages, and unfractionated, mature monocytes from human peripheral blood.[8–11] These studies support that osteoclasts originate from a myeloid cell origin.
Osteoclasts share several cellular and molecular properties of myeloid cells.[12] Myeloid cells comprise different cell types such as osteoclasts, granulocytes, monocytes, macrophages, and dendritic cells (DCs), which are found in various tissues in homeostatic conditions and circulate through the blood and lymphatic system. Myeloid cells have various functions and are recruited to the local sites where infection or tissue damage occurs. Monocytes are known to originate from the BM of a common myeloid progenitor (CMP) and are then released into the peripheral blood where they circulate for several days before entering tissues to replenish the tissue macrophage populations.[13–15] Circulating peripheral monocytes give rise to a variety of tissue-resident macrophages throughout the body as well as to specialized cells such as DCs and osteoclasts.[16,17] In addition to monocytes/macrophages, DCs also serve as a source of osteoclasts.[18–20] DCs are professional antigen-presenting cells that phagocytose, process, and present antigens to naïve T cells.[21] Although different types of myeloid cells can differentiate into osteoclasts, osteoclasts have unique functions that distinguish them from other myeloid cells; osteoclasts are the sole bone-resorbing cells that reside on bone and secrete acids and proteases to dissolve the bone matrix. Thus, the current paradigm proposes that the origin of osteoclasts may differ from that of other myeloid cells.
Molecular and cellular pathways of osteoclast differentiation have been well characterized and have provided a basis for therapeutic development for anti-resorptive therapies. Macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor (NF)-κB ligand (RANKL) are critical factors for osteoclast differentiation. Differentiation of osteoclast precursor cells (OCPs) into multi-nucleated osteoclasts is achieved through the activation of RANKL-responsive downstream factors and pathways such as NF of activated T cells 1 (NFATc1), c-Fos, and MYC.[1–4] Cell surface markers of mature osteoclasts are also well characterized. OCPs, the origin of osteoclasts, have been defined by many studies. Colony stimulating factor 1 receptor (CSF1R, also known as CD115) is a receptor for M-CSF and has been used as a surface marker for OCPs. Most studies have also relied on conventional surface markers for myeloid cells, including CD14, CD11b, CD117 (KIT), C-X3 -C motif chemokine receptor 1 (CX3CR1), and RANK. However, these markers used to define OCPs are also expressed in other myeloid cells. Thus, to date, defining OCP-specific markers has been limited.
Since OCPs are the origin of osteoclasts, OCPs can serve as a potential biomarker for detecting excessive bone loss and possibly as new therapeutic targets to combat bone loss in a wide range of bone diseases. Emerging evidence suggests the presence of distinct OCPs in pathological conditions, which also points to OCPs as a potential therapeutic target for pathological bone resorption. However, the contribution of distinct OCPs to pathological bone resorption remains to be solved. Thus, given the importance of osteoclasts in health and disease, the identification and classification of the specific combination of surface markers of OCPs have proven indispensable and will be required for the development of therapeutic interventions for pathological bone resorption.
THE DEVELOPMENTAL ORIGIN OF MURINE OSTEOCLASTS
In mammalian embryos, multiple precursors are involved in the production of distinct macrophages, which evolve in 3 temporally segregated waves.[22–24] Myelopoiesis begins in the extra-embryonic yolk sac through a stepwise process.[22–24] The first wave of the emergence of precursors is called primitive hematopoiesis, during which early erythro-myeloid progenitors (EMPs) are formed from the mesoderm in the blood islands of the extra-embryonic yolk sac at embryonic day 7 (E7). The primitive EMPs induce the production of erythroblasts and megakaryocytes, but they also differentiate into CSF1R+ yolk sac macrophages at E8.5. The second wave of hematopoiesis produces late EMPs at E8.25 from the hemogenic endothelium. CX3CR1 is a receptor for fractalkine,[25] and its expression, in combination with other markers, is used for defining monocyte subsets.[26] Late EMPs can either differentiate into CX3CR1+ yolk sac macrophages at E8.5, or they can colonize the fetal liver at E9.5 once the blood circulation has been established at E8.5. The last wave generates fetal HSC precursors at E10.5 in the aorta-gonad-mesonephros region that migrate into the fetal liver. The precursors differentiate into HSCs at E12.5 that colonize the fetal liver and later in the BM. HSCs mature and expand in the fetal liver. The fetal liver serves as a transient source of definitive hematopoiesis which is decreased and replaced by BM hematopoiesis.[13,27]
Increasing evidence suggests that cells of EMP origin and HSC lineage cells can form embryonic and postnatal osteoclasts and serve differential roles in osteoclastogenesis when osteoclasts are traced to their sources by different fate-mapping mouse models (Fig. 1).[28] Using Csf1r-Mer-iCre-Mer; Rosa26tdTomato fate mapping mice, Yahara et al. [28] revealed the contribution of EMP-derived monocytes/macrophages to producing neonatal osteoclasts.[29] The 80% of tartrate-resistant acid phosphatase (TRAP)+ osteoclasts in the P0 femur were derived from E8.5 CSF1R+ EMPs; however, they disappeared after 2 weeks. Moreover, genetic lineage tracing CX3CR1+ macrophages of EMP origin at E8.5 and E9.5 using CX3CR1creER;R26tdTomato fate mapping mice showed that not only do they contribute to neonatal osteoclasts, but the CX3CR1+TRAP+ double-positive population also survived in adult femurs at 6 months of age, supporting that EMP-derived macrophages are critical for the normal fetal skeleton and are survived for at least 6 months after birth. In addition, lineage tracing using Flt3cre;R26tdTomato fate mapping mice suggests that cell fusion between HSC-derived mononuclear cells and osteoclasts contributes to postnatal osteoclasts and maintains steady-state bone remodeling. Although the exact developmental origin of osteoclasts is still under debate, this study in which labeling precursors are induced at several time points during development suggests that CSF1R+ or CX3CR1+ cells derived from EMPs and fetal HSCs can serve as OCPs and contribute to the generation of embryonic and postnatal osteoclasts.
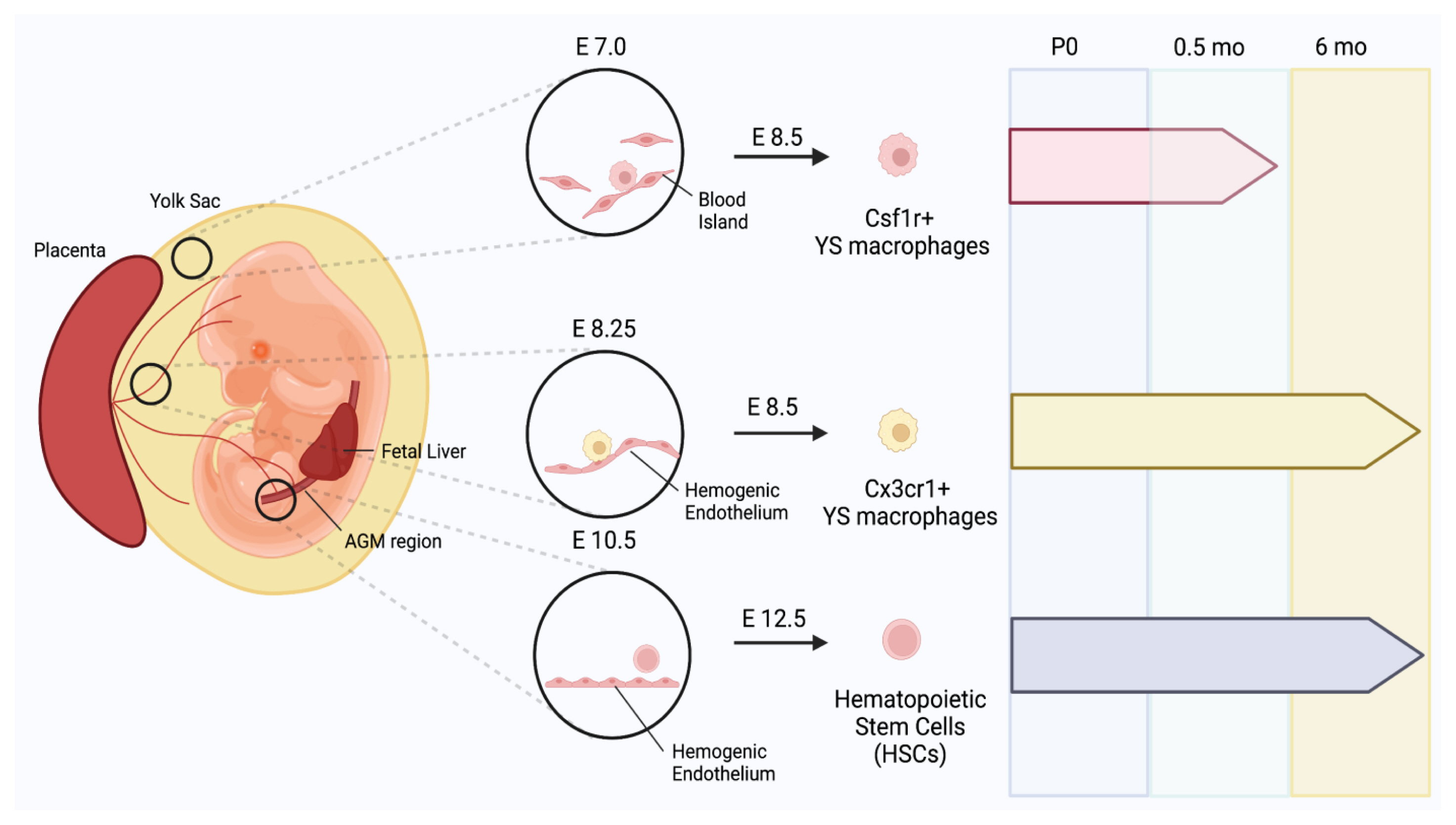
Emergence of yolk sac (YS) macrophage in mammalian embryos and their contributions to formation of neonatal osteoclasts. Mammalian embryos produce primitive erythro-myeloid progenitors (EMPs) at embryonic day 7 (E7). Primitive EMPs further give rise to colony stimulating factor 1 receptor+ (CSF1R+) YS macrophages at E8.5. Late EMPs are produced from the hemogenic endothelium at E8.25, inducing the emergence of CX3C motif chemokine receptor 1+ (CX3CR1+) YS macrophages at E8.5. Hematopoietic stem cell (HSC) precursors are also produced at E10.5 from the aorta-gonad-mesonephros (AGM) region, and they differentiate into HSCs at E12.5. CX3CR1+ macrophages mainly contribute to neonatal osteoclastogenesis. CSF1R+ YS macrophages were present at postnatal day 0 (P0). At P0.5 months (mo), CSF1R+ YS macrophages were only present in small amounts in the diaphysis but none were found in the metaphysis. CX3CR1+ macrophages that are differentiated in E8.5 and colonize the entire embryo at E9.5 were abundantly found in the P0 diaphysis and metaphysis. Unlike CSF1R+ macrophages, CX3CR1+ macrophages contribute to both neonatal osteoclastogenesis and bone remodeling in adult long bones. CX3CR1+ macrophages survive past 6 mo and reside in adult femurs. HSCs fuse with EMPs to produce multinucleated osteoclasts, which were found in P0 and adult femurs.
MURINE CIRCULATING OCP (cOCP)
Murine cOCPs represent a new frontier in the field of bone biology and in studying the lifespan of osteoclasts. They are defined as blood-borne cells with the capacity for osteoclastogenesis.[30] It is well accepted that cells with osteoclastogenic potential are circulating in the blood.[5,31–33] However, the identity of a murine circulator precursor cell remains elusive and its relation to BM progenitors is not well established.
The concept that cOCPs are fused with osteoclasts in bone injury sites has been solidified. The first discovery of this coordinated mechanism dates back to 1973 during which a cross-circulation experiment was conducted by parabiosis between mice protected from irradiation and injected with thymidine -3H and unlabeled thymidine and unprotected irradiated mice.[34] Thymidine -3H labeled multinucleated cells and osteoclasts were found in the fractured calluses of the unprotected mice, indicating the first possible contribution of blood-circulating precursors in osteoclastogenesis at the injury sites. With advancements in research technology, the role of cOCPs in perturbed microenvironments was further corroborated using reporter mice in the study by Novak, et al. [35]. CX3CR1+ cells were sorted from Cx3cr1GFP;TraptdTomato reporter mice via fluorescence-activated cell sorting analysis and sorted CX3CR1GFP cells were intravenously injected into wild-type mice prior to or after fracture. TRAPtdTomato+ multinucleated cells appeared in the bone repairing callus and newly forming bone. Consistent with these results, it has been shown that EMP-derived macrophages also infiltrate injury sites along with bone resident cells.[35] Yahara et al. [29] showed that EMP-derived CX3CR1+ cells could migrate to bone injury sites in a femoral drill hole model through the blood circulation. Taken together, these results support that cOCPs can contribute to osteoclast formation in bone injury sites (Fig. 2).
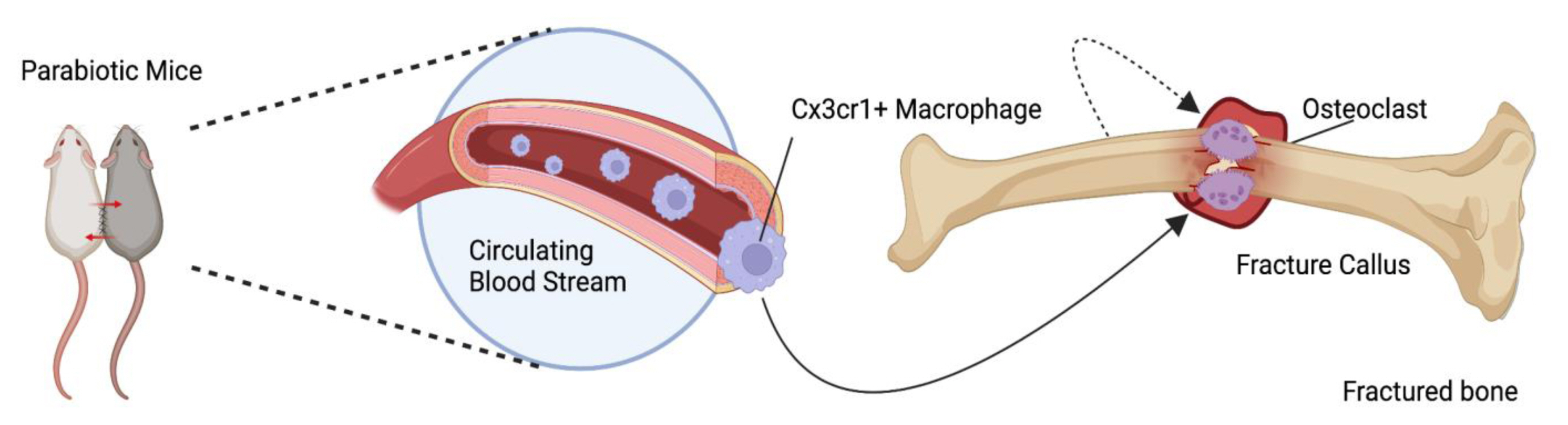
Circulating osteoclast precursor cells (OCPs) and their engraftment in bone injury. Circulating CX3C motif chemokine receptor 1+ (CX3CR1+) macrophages infiltrate injury sites of the disturbed bone marrow to repair the fractured callus and produce new bone. Circulating macrophages specifically infiltrate the fractured callus as well as the bone around the injury site to promote ossification. Despite the consensus surrounding the contribution of circulating OCPs in disturbed bone marrows, the definitive method by which hematopoietic stem cells interact with erythro-myeloid progenitors to create long-lived osteoclasts in normal homeostasis remains unclear.
Although osteoclasts have been considered terminally differentiated cells, recent studies suggest the presence of long-lived osteoclasts via constitutive fusion of cOCPs into pre-existing osteoclasts. In line with this, a parabiosis experiment by Jacome-Galarza et al. [36] proposes a gradual fusion of HSC-derived mononuclear progenitor cells with osteoclasts. The parabiosis between Csf1rcre;Rosa26LSL-YFP and Csf1rcre;Rosa26LSL-tdTomato mice showed that tdTomato signal intensity per YFP osteoclasts increased during parabiosis. Moreover, pulse labeling of the circulating nuclei incorporated with 5-ethynyl-2’-deoxyuridine and observing their fusion rates with osteoclasts suggest that EMP-derived long-lived osteoclasts are postnatally maintained by the serial acquisition of a new nuclei from HSC-derived mononuclear progenitors throughout steady-state bone remodeling. In contrast, Novak et al. [35] observed that HSC-derived mononuclear progenitors are unable to fuse with existing osteoclasts during normal homeostasis. Although it is evident that HSCs are a source of OCPs for generating osteoclasts in adulthood, the contribution of cOCPs to bone resident osteoclasts during homeostatic bone remodeling is still controversial.
MURINE BM OCP
Multiple studies have identified osteoclast progenitor populations from murine BM that are capable of differentiating into multinucleated osteoclasts following stimulation with M-CSF and RANKL.[9] However, the defined BM subpopulation designated as OCPs is largely varied among groups. Postnatal BM cells can differentiate into multiple progenitors of monocytes/macrophages, DCs, and osteoclasts.[28] These multi-potent progenitors are heterogenous and can advance downstream to result in the oligopotent progenitors of common lymphoid progenitors and CMPs.[37] CMPs give rise to megakaryocyte-erythrocyte progenitors (MEPs) and granulocyte-macrophage progenitors (GMPs).[38] GMPs produce common macrophage/osteoclast/DC progenitors (MODP). It has been shown that murine osteoclasts are derived from oligopotent MODP, B220−CD11blow/−c-Kit+c-Fms+ CD27+ Flt3+ cells.[39,40]
The osteoclastogenic potential of BM suspensions is screened by fractionating BM cells using multiple hematopoietic markers. Lee et al. [41] defined a distinct population of colony-forming cells as osteoclast colony-forming units in murine BM cells, adherent cell-depleted marrow, in the spleen, and in the day 14 fetal liver cultures cells. Muguruma and Lee [42] identified a distinct population of cells that gave rise to mononuclear cells with key features of osteoclasts in murine BM cultures after being stimulated by the conditioned medium of a bone-resorbing murine mammary carcinoma; upon further examination using cell surface markers, they also found that the early osteoclast activity was recapitulated by the fraction which is depleted of lineage-positive cells specific for murine B lymphocytes (CD45R, also known as B220), macrophages (CD11b), granulocytes (Gr-1), and erythroid cells (YW25.12.7). Arai et al. [39] found that BM CD11b−loCD115+CD117+ cells are enriched with high frequency of osteoclast precursor populations, although this population is also capable of differentiating into macrophages and DCs.[43] It was previously proposed that CD45R+ cells contain OCPs.[44] Katavić et al. [44] found that purified BM CD45R+ cells are OCPs that respond to estrogen, and ovariectomy was found to increase CD45R+ OCPs. However, Jacquin et al. [45] demonstrated that sorted CD45R+ BM cells were unable to differentiate into osteoclasts in vitro when stimulated with M-CSF and RANKL. B220−CD3−CD45R−CD11b−/low CD115+CD117+ cells from total BM cells show early osteoclastogenic activity. CD117 expression was downregulated as osteoclasts matured and were used to mark OCPs. However, CD117− cells were also able to differentiate into osteoclasts albeit with lower efficiency. Charles et al. [46] identified that a sorted B220−CD3−CD11b−/low Ly6Chi population is highly enriched for osteoclast formation when cultured in M-CSF and RANKL, which increased following the induction of arthritis. These cells were distinct from other BM myeloid precursor populations, demonstrating anti-CD11c activity, and were also positive for CX3CR1. Das et al. [47] showed that Ly6Chi and Ly6Cint monocytes in BM are a main source of osteoclasts. Intriguingly, several groups also demonstrated that distinct BM OCP populations give rise to osteoclasts in pathological conditions involving increased osteoclast activity and enhanced bone resorption (please see section 5). Taken together, multiple populations in BM have the capacity to differentiate into osteoclasts under M-CSF and RANKL or in co-culture with stromal cells (Fig. 3).

Bone marrow (BM) and circulating osteoclast (OC) precursor cell (OCP) surface markers in murine and human models. (Left panel) CD11b−loCD115+CD117+ BM cells have high frequency of OCP populations and can differentiate into OCs upon stimulation with macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor-κB ligand (RANKL). Further fractionation of BM cell populations has identified a population of B220−CD3−CD45R−CD11b−/lowCD115+CD117+ cells that show early osteoclastogenic activity. CD11b−/lowLy6Chi cells in murine BM are enriched for osteoclast information upon co-culture with M-CSF and RANKL and are greatly increased following the induction of arthritis. In murine models, circulating CX3C motif chemokine receptor 1+ cells have been shown to migrate to the bone and become mature OCs. Parabiosis experiments between Csf1rcre;Rosa26LSL−YFP and Csf1rcre;Rosa26LSL-tdTomato were able to demonstrate that tdTomato+ OCs are replenished by circulating YFP+ precursor cells. (Right panel) Human blood monocytes are segregated based on their cell surface expression of CD14 and CD16 into non-classical (CD14+CD16+), classical (CD14+CD16−), and intermediate (CD14++CD16+) monocytes; all three subsets can differentiate into OCs. In human BM, the presence of CD117 and FLT3 and absence of CD11b are used to define human hematopoietic stem cells, which contains the common progenitor of GMODP in addition to MODPs.
HUMAN OCP
Human OCPs have been documented to a lesser extent than murine OCPs. Both human BM cells and peripheral CD14+ monocytes can differentiate into osteoclasts in vitro and are considered as a source of OCPs. In this section, we will review the current understanding of human OCPs.
1. Human blood OCP
Fujikawa et al. [48] provided the first evidence showing that human blood mononuclear cells contain OCPs; in vitro co-culture of peripheral blood mononuclear cells with osteoblastic cells formed osteoclasts. Monocytes were initially defined by their distinct morphology and later by expression of cell surface markers including CD14, a co-receptor for toll-like receptor 4.[17] Several studies demonstrated that among blood mononuclear cells, the culture of unfractionated peripheral-blood CD14+ monocytes with M-CSF and RANKL is sufficient to induce osteoclast formation. Peripheral-blood monocytes show morphological heterogeneity, such as variability in size, granularity, and nuclear morphology [15] and transcriptomic heterogeneity.[49] The development of flow cytometry analysis enables the separation of heterogeneous monocyte subsets by applying knowledge of cell surface markers.[50] Human blood monocytes have been segregated into 3 subsets based on their cell-surface expression of CD14 and CD16 (also known as FcγRIII) - classical CD14+CD16− monocytes, non-classical CD14dimCD16+ monocytes, and intermediate CD14+CD16+ monocytes.[50,51] Several studies demonstrated that these 3 subsets based on their expression of CD14 and CD16 exhibit distinct cellular phenotypes and functional differences in migration, protein expression, gene expression, cytokine secretion, and differentiation potential into macrophages, DCs, or osteoclasts.[52–55] However, which subset serves as an OCP population remains inconclusive. Sprangers et al. [55] showed that all 3 subsets could differentiate into osteoclasts in vitro. Intriguingly, the proportion of non-classical and intermediate monocytes in synovial fluids or blood varies among inflammatory diseases.[54] Of note, most of the studies that investigate osteoclasts report non-classical and intermediate monocytes as CD16+ monocytes. CD16+ monocytes are observed to be increased in the blood of psoriatic arthritis (PsA) patients and in the BM of multiple myeloid patients.[51,56,57] Moreover, CD16+ monocytes are more prone to differentiate into osteoclasts compared to other subsets of monocytes. In contrast, several other studies demonstrated that osteoclasts were mainly derived from CD14+CD16− classical monocytes but not from CD16+ monocytes in healthy donors and patients with RA.[58–60] Komano et al. [60] showed that CD14+CD16− monocytes differentiate into osteoclasts. However, CD14+CD16+ monocytes are able to respond to RANKL to produce tumor necrosis factor (TNF) and interleukin-1 (IL-1) but are unable to differentiate into osteoclasts.[60] We also observed that CD14+CD16− monocytes are a main source of osteoclastogenesis (Kaneko et al. unpublished observations).
2. Human BM OCP
Osteoclasts are also derived from human BM cells.[61–66] BM cells are heterogenous, and most of our understanding of HSC hierarchical organization results from murine studies (please see section 3). As described above, murine osteoclasts, macrophages, and DCs differentiate from MODP. [39,40] However, equivalent HSC hierarchical organization on the origin of osteoclasts has not been fully described. The presence of CD34 has been used as a marker of human HSCs.[67] Xiao et al. [66] sorted CD34+c-KIT+FLT3+ IL3αhigh cells from pediatric BM by flow cytometry as MODP. By analogy with the mouse MODP, the presence of c-KIT (CD117) and FLT3 (CD135), and the absence of CD11b are used to define human MODP.[39,68] These markers are expressed on the murine common DC progenitor and upstream precursors, where high FLT3 levels distinguish the mouse lympho/myeloid precursor from the MEP.[69,70] CD34+c-KIT+ FLT3+ cells, constitute less than 0.5% of total pediatric BM and have proved to exhibit osteoclastogenic potential. CSF1R mRNA was expressed in CD34+c-KIT+FLT3+ precursor cells, but CSF1R protein was not detected on the cell surface. Further investigation of the surface antigens of CD34+c- KIT+FLT3+ precursor cells identified that this cell is also Lin−CD7−CD10−CD38+CD45RA+. Among CD11b−CD34+c-KIT+FLT3+ BM cells, IL3Rα expression levels discriminated between 2 consecutive stages of macrophage, osteoclasts, and DC differentiation. The IL3Rαlow subset of this BM population constitutes the common progenitor of granulocyte, macrophage, osteoclasts, and DCs. The IL3Rαhigh subset lies downstream from the GMODP and constitutes the newly identified human MODP, oligopotent for macrophage, osteoclasts, and DC differentiation.
Taken together, although these studies suggested that both human BM cells and blood cells contain osteoclast precursor populations (Fig. 3), defined human OCPs are heterogenous. Thus, further characterization of OCPs in humans can provide insight into the regulation of human osteoclasts.
MODULATION OF OCP IN PATHOLOGICAL CONDITIONS
1. Chronic inflammation
Elevated osteoclast activity is a major cause of inflammatory bone erosion in chronic inflammatory diseases.[4,71] The concept that osteoclasts in inflammatory conditions are derived from distinct OCPs that may differ from OCPs under physiological conditions has recently emerged.
1) Arthritis-associated osteoclastogenic macrophages (AtoMs)
Increased number and enhanced activity of OCPs were observed in murine inflammatory arthritis models. Seeling et al. [72] showed that osteoclasts are derived mainly from Ly6Chi inflammatory monocytes in inflammatory conditions. Charles et al. [46] demonstrated that Ly6ChiCD11b−/lo OCPs increased following the induction of arthritis in the SKG arthritis model. Ly6ChiCD11b−/lo OCPs expressed CX3CR1.[73] Consistently, Ibáñez et al. [74] also identified CX3CR1 as a surface marker of inflammatory osteoclasts in a murine inflammatory colitis model. While CX3CR1+ macrophages were observed in the synovial lining and restricted the inflammatory reaction by functioning as an internal immunological barrier,[75] Hasegawa et al. [76] showed that osteoclasts are formed only from CX3CR1+ cells in inflamed synovium. This study further identified murine CX3CR1hiLy6CintF4/80+ I-A+/I-E+ macrophages (called AtoMs) as distinct OCPs in the inflamed synovium from a collagen-induced arthritis model. This study proposed that CX3CR1lo Ly6Chi cells (known as inflammatory monocytes) in the blood migrate to the inflamed synovium and transition into CX3CR1hi Ly6Cint cells. A subset of CX3CR1hi Ly6Cint cells are further defined by the expression of cell surface markers such as F4/80, I-A/I-E, CD80/86, and CD11c. This subset of cells, which were named AtoMs, has prominent osteoclastogenic ability especially under combined stimulation with RANKL and TNF-α. Furthermore, they also found that forkhead box M1 (FOXM1) is a key regulator of AtoMs by transcriptional profiling. FOXM1 deletion inhibited the osteoclastogenic potential of AtoMs, and FOXM1 inhibition in synovial CX3CR1+HLA-DrhiCD11c+ CD80−CD86+ cells from RA patients suppressed osteoclastogenesis. This study suggests the existence of a distinct OCP subset that may be responsible for bone erosion in inflammatory conditions.
2) Inflammatory OCPs (iOCPs)
Meirow et al. [77] identified 2 distinct subsets of OCPs in chronic inflammation and suggested the emergence of a distinct OCP subset (named iOCPs) during chronic inflammation. Ly6ChiCD11bhi iOCPs are induced in chronic inflammation models, while Ly6ChiCD11blo homeostatic OCPs (hOCPs) remain unchanged in inflammatory conditions. Functional and proteomic analyses further characterized that iOCPs are quickly expanded and generate highly active osteoclasts in chronic inflammatory conditions such as inflammatory arthritis. However, iOCPs were rarely present and had low osteoclastogenic potential under homeostatic conditions, whereas hOCPs were abundant and manifested high osteoclastogenic potential under physiological conditions. hOCPs formed osteoclasts with low activity under inflammatory conditions. Osteoclasts derived from iOCPs expressed higher levels of resorptive and metabolic proteins than those generated from hOCPs. Furthermore, TNF-α and S100A8/A9 proteins are important regulators of the iOCP response during chronic inflammation. Madel et al. [78] demonstrated the heterogeneity of iOCPs by defining 2 groups of iOCPs – CX3CR1+ iOCPs and CX3CR1− iOCPs. Future investigation is needed to identify if iOCPs are responsible for inflammatory bone loss in humans.
3) Human CD14+CD16+ cells
In humans, the expansion of the CD16+ monocytes in many different types of diseases, mostly in infection or inflammatory conditions, has been described.[51,79–82] This subset is generally termed “pro-inflammatory” monocytes because of their ability to produce high amounts of TNF-α and IL-1β.[60,83,84] Table 1 summarizes a list of inflammatory diseases and malignant tumor diseases related to bone metabolism and the frequencies of each of the 3 monocyte subsets as reported in the literature.

Three subsets of cluster of differentiation 14+ monocytes in patients with inflammatory diseases and multiple myeloma
(1) RA
RA is a chronic systemic, inflammatory and autoimmune disease of unknown etiology characterized by inflammation of synovitis, hyperplasia of synovial tissue, structural destruction of cartilage with pannus formation and bone erosions preferentially surrounding peripheral synovial joints.[85] Of the 3 subsets of monocytes described above, intermediate monocytes (CD14++CD16+) are greatly increased in number and quantity in the peripheral blood and synovium of RA patients.[86–90] Several reports show that the level of classical monocytes is increased in RA patients compared to that of normal healthy controls,[86–90] while other reports show that the level of non-classical monocytes is increased in RA patients with no significant changes in the level of classical monocyte subsets than the healthy controls.[89] There appears to be a developmental relationship among the 3 monocyte subsets in RA patients. Intermediate monocytes produce high concentrations of the pro-inflammatory cytokines TNF-α, IL-1β, and IL-6 in the RA synovium, suggesting that they can differentiate into inflammatory macrophages[91] which promote local synovial inflammation.[92] These CD14++CD16+ monocytes activate Th17 cells via direct cell contact in the inflamed synovial tissue and promote the expansion of Th17 cells.[92,93]
(2) PsA
PsA is an inflammatory joint disease that can be distinguished from RA based on unique clinical features, the absence of rheumatoid factor, and characteristic radiographic findings. Patients with PsA frequently develop focal inflammation at multiple sites, including the skin, joints, and tendon-insertion sites or entheses.[94] In patients with PsA, the CD16+ monocytes exhibit a preferential ability to differentiate into osteoclasts. However, in healthy controls, the classical monocytes harbor the highest propensity to differentiate into osteoclasts.[56] This led to the suggestion that the CD16+ cells in PsA patients may have derived from the classical subset since they exhibit similar properties – in this case, the ability to become osteoclasts. It is not known what induces CD16 expression in classical monocytes, but M-CSF and/or RANKL are likely candidates in this disease setting since these factors together in vitro can induce CD16 expression in monocytes.
(3) Multiple myeloma (MM)
MM can modify their surrounding immune and bone microenvironments, interfere with immune surveillance, and ultimately lead to bone resorption and lysis via direct local cell-to-cell and cytokine-mediated interactions between monocyte/macrophages, stromal cells, osteoclasts, and osteoblasts.[95,96] Elevated levels of multiple serum inflammatory cytokines in MM patients have been associated with increased bone resorption markers. Damasceno et al. [57] reported the distribution of monocyte subsets in the blood of MM patients and their association with serum inflammatory and bone-associated biomarker profiles in a subset of the patients. Classical monocytes were diminished in the blood of MM patients, while intermediate and non-classical monocytes were significantly increased in BM of patients with MM.[97] Increased serum IL-8 was characteristic of MM, and higher serum IL-6, RANKL, and bone alkaline phosphatase concentrations together with decreased counts of FcɛRI+ classical monocyte were restricted to MM presenting with bone lesions. These results provided new insights into the pathogenesis of MM and the potential role of FcɛRI+ classical monocytes in normal bone homeostasis.[57] However, the role of CD16+ monocytes in bone lesion of MM remains controversial. While Bolzoni et al. [98] showed that CD16+ monocytes from patients with MM have higher osteoclastogenic potential than CD14+CD16− classical monocytes in ex vivo cultures, Petitprez et al. [99] reported that non-classical CD16+ monocytes were not correlated with bone lesions in MM.
PERSPECTIVES
In this review, we highlight key features of OCPs. Although it is not easy to draw clear lines between osteoclast precursors and other myeloid progenitor cells, OCPs seem to show several distinct features and should be classified as a separate population. However, the understanding of OCPs is still at an early stage and further study is warranted. Several open questions for OCP biology still need to be resolved. First, it remains unclear if only one population of OCPs exists in the body. A recent study showed that myelopoiesis is altered when mice are exposed to inflammatory conditions.[100] Osteoclast formation and activity are also altered by different environmental cues – either systemic or local signals. Elevated osteoclast activity and number have been implicated in the case of inflammatory bone loss in chronic inflammatory diseases. Emerging evidence supports that osteoclasts in inflammatory conditions can be derived from iOCPs that are different from hOCPs. However, it still remains unclear whether pathogenic osteoclasts are derived from a different source of OCPs that are generated only in pathological conditions. It is also possible that iOCPs and hOCPs originate from the same OCPs, but iOCPs are differentially reprogrammed under pathological conditions. A better understanding of cell surface markers of heterogenous OCPs will address this question. Another question is whether osteoclasts across different bone sites come from the same OCPs. It will be valuable to compare OCPs from different bone sites. Thirdly, osteoclasts reside in bone; however, it has not been fully explored how different subsets of OCPs are recruited to the bone. Chemokines and M-CSF can be potential regulators for the recruitment of OCPs since those factors play important roles in recruiting macrophages.[101] Although CX3CR1 is a chemokine receptor and a marker for iOCPs, the function of CX3CR1 is not required for bone loss in osteoporosis and arthritic bone erosion.[78] In summary, OCPs can be major therapeutic targets of interventions for bone diseases. However, the molecular and cellular properties of OCPs which are unique to OCPs are not well elucidated. Given the ability of osteoclasts to resorb bone and maintain calcium in the body, a better understanding of the nature and heterogeneity of OCPs and molecular pathways responsible for the recruitment of OCPs to bone can be explored as potential targets for osteoclast-specific therapies.
Acknowledgements
The figures are generated by Biorender.
Notes
Ethics approval and consent to participate
Not applicable.
Conflict of interest
No potential conflict of interest relevant to this article was reported.