Pathobiology of Paget's Disease of Bone
Article information
Abstract
Paget's disease of bone is characterized by highly localized areas of increased bone resorption accompanied by exuberant, but aberrant new bone formation with the primary cellular abnormality in osteoclasts. Paget's disease provides an important paradigm for understanding the molecular mechanisms regulating both osteoclast formation and osteoclast-induced osteoblast activity. Both genetic and environmental etiologies have been implicated in Paget's disease, but their relative contributions are just beginning to be defined. To date, the only gene with mutations in the coding region linked to Paget's disease is sequestosome-1 (SQSTM1), which encodes the p62 protein, and these mutations lead to elevated cytokine activation of NF-B in osteoclasts but do not induce a "pagetic osteoclast" phenotype. Further, genetic mutations linked to Paget's appear insufficient to cause Paget's disease and additional susceptibility loci or environmental factors may be required. Among the environmental factors suggested to induce Paget's disease, chronic measles (MV) infection has been the most studied. Expression of the measles virus nucleocapsid gene (MVNP) in osteoclasts induces pagetic-like osteoclasts and bone lesions in mice. Further, mice expressing both MVNP in osteoclasts and germline mutant p62 develop dramatic pagetic bone lesions that were strikingly similar to those seen in patients with Paget's disease. Thus, interactions between environmental and genetic factors appear important to the development of Paget's disease. In this article we review the mechanisms responsible for the effects of mutant p62 gene expression and MVNP on osteoclast and osteoblast activity, and how they may contribute to the development of Paget's disease of bone.
INTRODUCTION
Paget's disease of bone is a late-onset skeletal disease affecting 2-5% of Caucasians over 55 years old that is characterized by highly localized areas of increased bone resorption accompanied by exuberant, but aberrant, new bone formation that results in bony expansion and structural weakness of the involved bone. The primary lesion in Paget's involves the formation of abnormal osteoclasts (OCL) which express a "pagetic phenotype" that includes increased OCL number and size, increased nuclei/OCL, increased responsivity of the OCL precursors to OCL-inducers receptor activator of nuclear factor (NF)-kappaB (κB) ligand (RANKL), tumor necrosis factor (TNF)-α, and 1,25-dihydroxy-vitamin D3 (1,25-[OH]2D3), increased production of the cytokine interleukin-6 (IL-6), and altered expression of signaling molecules and transcription factors (Fig. 1).[1] Further, the pagetic OCL also express increased levels of coupling factors which drive aberrant bone formation.[2] The excessive focal bone formation in Paget's results in the generation of weak woven bone, with collagen fibers laid down in an irregular mosaic pattern, rather than normal lamellar bone.[3] The pagetic bone that is formed can bow and result in bone deformity or fracture, skull thickening, bone pain, secondary osteoarthritis, and nerve root compression.[4] Paget's disease represents the most exaggerated example of OCL-osteoblast "coupling" with both bone resorption and bone formation markedly increased. Therefore, Paget's disease provides an important paradigm for understanding the molecular mechanisms regulating both aberrant OCL formation and OCL-induced osteoblast activity.
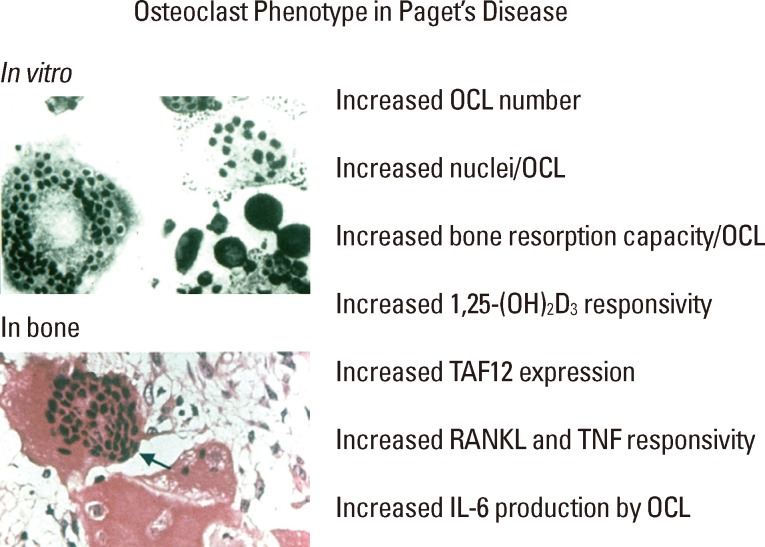
Characteristics of the pagetic osteoclasts phenotype. OCL, osteoclasts; 1,25-(OH)2D3, 1,25-dihydroxy-vitamin D3; TAF12, TAF12 RNA polymerase II, TATA box binding protein (TBP)-associated factor, 20kDa; RANKL, receptor activator of NF-kappaB ligand; TNF, tumor necrosis factor; IL-6, interleukin-6.
The etiology of Paget's disease is complicated and unclear. Both genetic and environmental etiologies have been implicated in the pathophysiology of Paget's disease. [1,5] The disease mainly affects the elderly, with age of initial diagnosis usually in the late 60's, and is approximately 2-fold more common is men than in women.[6] The incidence of Paget's disease varies widely between geographic regions, with the highest prevalence in people of Anglo-Saxon origin in Great Britain, Australia, New Zealand and North America, followed by Western and Southern European countries, whereas it is rare in Scandinavia, Africa, and Asia.[5] Between 15-30% of Paget's patients have a family history of the disorder with an autosomal dominant pattern of inheritance, suggesting a genetic predisposition for Paget's disease.[7,8]
Genetic contribution
To date, the only gene with mutations in the coding region that has been linked to Paget's disease is sequestosome-1 (SQSTM1) on 5q35, which encodes the p62 protein, a scaffolding protein involved in mediating cytokine signaling and that can serve as a cargo adaptor for polyubiquinated proteins in both proteasomal degradation and autophagy.[9,10] All 28 p62 mutations identified in Paget's patients result in loss of function of the C-terminal ubiquitin association (UBA) domain and lead to elevated cytokine activation of NF-κB and a pagetic OCL phenotype. The most commonly found mutation C1215T causes an amino acid substitution of proline (P) to leucine (L) at codon 392 (p62P392L) and is found in 10% of sporadic and 30% of familial Paget's patients.[11,12,13] Several studies have indicated that Paget's disease kindreds with p62 mutations show incomplete penetrance (15-20% of carriers fail to develop the disease) and variability of disease severity (age of onset, number of affected sites) among affected members with the same mutation.[14,15,16] Further, although the trends are not identical in all regions, Paget's disease has been reported to be decreasing in prevalence and severity over the past 20-30 years.[17,18,19,20] This supports the hypothesis that additional factors may be required to cause the disease, either additional susceptibility loci or environmental factors, and further suggests an interaction between genetic factors and environmental triggers that may be changing.
Genome-wide studies have recently identified 7 loci with genetic polymorphisms (single nucleotide polymorphisms, SNPs) that have been linked to susceptibility to develop Paget's disease and together account for 13% of the familial risk of Paget's disease.[21,22,23,24,25] These regions contain 8 genes (1 region has two genes) that are intriguing as they have either known or theoretically likely effects on OCL functions: 1p12.3-macrophage colony stimulating factor (M-CSF; CSF1), 18q21.33-RANK (TNFRSF11A), 8q22.3-dendritic-cell-specific transmembrane protein (DC-STAMP; TM7SF4), 10p13-optineurin (OPTN), 7q33-nucleoporin 205 kDa (NUP205), 14q32-Ras and Rab interactor 3 (RIN3), 15q24-promyelocytic leukemia (PML) and golgin A6 family, member A (GOLGA6A). M-CSF and RANK are known to be involved in regulating OCL differentiation, and DC-STAMP is important for OCL fusion and multinucleation.[26,27] Roles in OCL differentiation or function for optineurin, NUP205, RIN3, PML, and GOLGA6A have not yet been defined. However, optineurin is a IκB kinase γ (IKKγ; also known as NEMO) homolog and can regulate NF-κB activation as well as serve as a specific cargo-adapter for autophagy,[28] functions that suggest that it could have a role in regulating OCL. Nucleoporin205 is a component of the nuclear pore complexes which regulate transport across the nuclear membrane,[29] but what role this might play in OCL or specifically in Paget's disease is unclear. RIN3 interacts with small GTPases such as Ras and Rab,[30] key regulators of OCL function and vesicular trafficking.[31] The PML protein has been reported to be involved in transforming growth factor (TGF)-β signaling,[32] although it has not been examined in the context of TGF-β regulation of bone metabolism. The GOLGA6A gene encodes a golgin family member. These coiled-coil proteins associate with the Golgi apparatus and have a role in membrane fusion as well as serve as structural supports for the Golgi cisternae. Mutations in other members of the golgin family can cause lethal skeletal dysplasia[33] and a severe form of osteoporosis.[34] However, only one of these Paget's susceptibility polymorphisms is in the coding region of a candidate gene (rs5742915 results in a phenylalanine to leucine amino acid change at codon 645 of PML), and it is not known how any of these susceptibility polymorphisms impact the expression and function of the associated candidate genes.
Environmental contribution
A number of environmental factors have been suggested as possible triggers for Paget's disease. These include dietary deficiencies (calcium, vitamin D), stress from repetitive use of affected bones, exposure to environmental toxins, rural lifestyle or animal exposure, and chronic infection with the paramyxoviruses measles virus (MV), canine distemper virus (CDV), and respiratory syncytial virus (RSV).[35] More than 30 years of studies have supported the hypothesis that Paget's disease may result from a chronic paramyxoviral infection. Ultrastructural studies by Rebel and coworkers[36] revealed that nuclear and less commonly, cytoplasmic inclusions resembling paramyxoviral nucleocapsids were present in pagetic OCL. In addition, Mills et al.[37] used immunohistochemistry to show that antigens from RSV and MV nucleocapsids were detected in OCL from Paget's patients, but not in OCL from with other bone diseases. Similarly, CDV nucleocapsid protein was reported in 11/25 English Paget's patients by Gordon and coworkers using in situ hybridization.[38] Further, using highly sensitive in situ polymerase chain reaction (PCR) techniques, Mee and colleagues[39] found CDV nucleocapsid transcripts expressed in OCL from 12/12 English Paget's patients. Kurihara and coworkers found using real-time PCR that ~70% of Paget's patients carrying the p62P392L mutation had marrow cells that were positive for expression of MV nucleocapsid protein (MVNP).[40] However, other groups using an array of techniques have been unable to detect MV or CDV in cells from Paget's patients,[35] making the role of a chronic paramyxoviral infection in Paget's disease controversial.
Evidence for a pathophysiological role for MV in the abnormal OCL activity in Paget's disease was first provided by Kurihara et al.[41] They demonstrated that OCL formed from normal human OCL precursors (CFU-GM) transduced with retroviral vectors expressing the MVNP gene displayed many of the abnormal characteristics of pagetic OCL. Furthermore, infecting canine bone marrow (BM) cells with CDV resulted in the development of pagetic-like OCL.[42] Similarly, when MV was used to infect BM cells from transgenic mice expressing the CD46 cellular receptor for MV, the OCL formed in vitro expressed aspects of a pagetic OCL phenotype.[43] Importantly, Kurihara and colleagues[40] recently showed that antisense knockdown of MVNP expression in MVNP-positive OCL precursors from Paget's patients carrying p62P392L resulted in loss of the pagetic OCL phenotype, indicating that p62P392L appears insufficient to induce a pagetic phenotype in OCL from p62P392L patients, and that MVNP is a required cofactor in at least a subset of Paget's disease patients. Intriguingly, the MVNP-expressing OCL precursors from Paget's patients carrying p62P392L were hyper-responsive to 1,25-(OH)2D3, expressed increased TAF12 RNA polymerase II, TATA box binding protein (TBP)-associated factor, 20kDa (TAF12), a VDR coactivator, and formed hyper-nucleated OCL in vitro, whereas, the MVNP-negative OCL precursors from Paget's patients carrying p62P392L did none of these, although the patients had pagetic lesions in vivo. These results suggest that possibly a second genetic mutation is required for the development of Paget's disease in the MVNP-negative patients, or possibly another environmental trigger such as RSV or CDV.
Mouse models
Further demonstration of the multifactorial nature of Paget's disease comes from animal models. Kurihara and colleagues[44] reported that both transgenic mice expressing human p62P392L specifically in the OCL lineage driven by the tartrate-resistant acid phosphatase (TRAP) promoter (TRAP-p62P392L mice) and knock-in mice expressing the murine equivalent (p62P394L) in the germline (p62P392L KI mice)[40] failed to develop pagetic bone lesions in the lumbar vertebrae or have characteristic abnormal pagetic OCL, although they did have increased OCL numbers. Further, in vitro studies revealed that the OCL precursors from these mice did not display increased responsivity to 1,25-(OH)2D3 or hyper-multinucleation, although they did have increased responsivity to RANKL and TNF-α. In contrast, Daroszewski et al.[45] also recently analyzed a p62P394L KI mouse model and reported that they found small pagetic lesions in the long bones, but not in the vertebrae. In addition, they observed OCL hypernucleation both in the lesions and after in vitro OCL formation. It is unclear why the two similar mouse models yielded different results, but this suggests that the capacity of mutant p62 to induce pagetic lesions in vivo is variable and that there may have been an additional environmental factor involved. Strikingly, Kurihara et al.[46] reported that 29% of transgenic mice with MVNP expression targeted to the OCL lineage (TRAP-MVNP mice) developed localized bone lesions in the L1-4 vertebrae that are similar to lesions seen in patients with Paget's disease. More recently, these investigators reported that mice expressing both TRAP-driven MVNP and the p62P394LKI develop dramatic pagetic bone lesions that were strikingly similar to those seen in patients with Paget's disease.[40] The bone marrow derived OCL precursors isolated from both models expressing MVNP exhibited pagetic OCL characteristics upon differentiation with either RANKL, TNF-α, or 1,25-(OH)2D3.[40,46] However, OCL precursors from mice expressing only TRAP-p62P392L or p62P394LKI formed increased OCL, but did not exhibit all aspects of pagetic OCL, such as hyper-multinucleation or increased IL-6 expression.[40,44] It is important to note that in all these mouse models, aging was still required with bone lesions usually not seen until one year of age, analogous to Paget's disease of bone occurring only predominantly in older patients carrying genetic susceptibilities, and that the lesions are focal. The latter may account for the apparent incomplete penetrance as only a limited set of bones were analyzed in the mouse models.
Role of IL-6
Paget's patients have elevated IL-6 levels in their marrow plasma and peripheral blood, and their OCL express high levels of IL-6.[40,47] Menaa et al.[48] reported that IL-6 blocking antibodies decreased the RANKL responsivity of pagetic marrow to normal levels, whereas addition of IL-6 to normal marrow enhanced RANKL responsivity. Similarly, anti-IL-6 antibodies reduced OCL formation from MVNP-expressing mouse BM cells.[46] Importantly, Kurihara et al.[40] reported IL-6 deficiency in TRAP-MVNP mice abrogates the development of pagetic OCL both in vivo and in vitro, revealing that many of the effects of MVNP seen in these Paget's disease mouse models above were moderated by IL-6. In contrast, OCL from p62P392L KI mice do not express elevated IL-6 levels and these mice do not have pagetic lesions. Therefore, the high levels of IL-6 induced by MVNP in OCL precursors are essential for the formation of pagetic OCL and bone lesions.
To examine if over-expression of IL-6 by itself is sufficient or can collaborate with mutant p62 to generate the complete Paget's bone phenotype, Teramachi and colleagues[49] generated TRAP-driven IL-6 transgenic mice (TIL-6) and bred them to the p62P392L KI mice to create p62KI/TIL-6 mice. The TIL-6 mice produce IL-6 at levels comparable to TRAP-MVNP mice. Greater numbers of OCL formed in response to 1,25-(OH)2D3 from p62KI/TIL-6 OCL precursors than either p62KI or TIL-6 OCL precursors. Although p62KI/TIL-6 had increased numbers of nuclei/OCL, the nuclear number was lower than in TRAP-MVNP mice. While p62KI/TIL-6 mice bones had increased OCL numbers per surface compared to wildtype (WT) mice, no pagetic OCL or lesions were detected in vivo, and µqCT analysis of bones from these mice did not reveal any significant differences between p62KI/TIL-6 and WT mice. Thus, increased IL-6 expression in OCL from p62KI mice contributes to increased responsivity to 1,25-(OH)2D3 and increased OCL numbers, but is not sufficient to induce Paget's-like OCL or bone lesions in vivo.
Mechanisms of increased responsivity of pagetic OCL precursors to OCL inducers
A key feature of pagetic OCL precursors is that they are sensitive to lower levels of osteotropic factors than normal precursors. In marrow cultures from patients with Paget's disease, OCL are formed in response to 10-fold lower concentrations of the cytokines RANKL[1,50] and TNF-α[44] than normal cells. Similarly, pagetic OCL precursors form OCL at physiological (10-11 M) 1,25-(OH)2D3, whereas normal OCL precursors form OCL at pharmacological (10-8 M) 1,25-(OH)2D3.[41,50] Both MVNP and mutant p62 make OCL precursors more sensitive to RANKL and TNF-α, but only MVNP sensitizes the cells to 1,25-(OH)2D3.[40,44,46] Of note, as mentioned earlier, Kurihara et al.[40] found that 1,25-(OH)2D3 hypersensitivity correlated with MVNP expression in OCL precursors from Paget's patients carrying p62P392L, and this was lost when MVNP was knocked-down.
OCL precursors from Paget's patients have increased expression of transcription initiation factor TFIID subunit 12 (TAF12; formerly known as TAFII-17), a member of the TFIID transcription initiation complex. Importantly, TAF12 also acts as a vitamin D receptor (VDR) coactivator enhancing the ability of VDR to respond to lower amounts of its ligand 1,25-(OH)2D3.[51] Therefore, the elevated TAF12 levels in pagetic OCL precursors sensitizes the cells to lower 1,25-(OH)2D3 levels. The 1,25-(OH)2D3 hypersensitivity of OCL precursors correlated with increased TAF12 and the presence of MVNP. Chromatin immunoprecipitation demonstrated TAF12 binding to a VDR target gene, cytochrome P450, family 24, subfamily A, polypeptide 1 (Cyp24A1), promoter containing two functionally important VDR binding sites.[52] In addition, TAF12 was reported to interact with the bZIP activating transcription factor 7 (ATF7) and potentiate ATF7 driven genes.[52] MVNP expression elevated ATF7 protein levels compared to normal OCL precursors independent of 1,25-(OH)2D3. Coimmunoprecipitation studies found that TAF12 interacts with ATF7 in MVNP-expressing OCL precursors. Further, ATF7 knockdown in MVNP-expressing OCL precursors decreased 1,25-(OH)2D3 induction of the TAF12-VDR target gene Cyp24A1. Thus, ATF7 increases the TAF12 contribution towards the 1,25-(OH)2D3 hypersensitivity of pagetic OCL precursors (Fig. 2A). TAF12 over-expression by retroviral transduction in normal human OCL precursors and in transgenic mouse OCL precursors from TRAP-TAF12 mice resulted in OCL precursors with increased sensitivity to 1,25-(OH)2D3 and led to the formation of OCL at lower levels of 1,25-(OH)2D3 than WT cells.[53,54] However, these OCL failed to exhibit multi-nucleation or increased sensitivity to RANKL. Further, the TRAP-TAF12 mice did not develop pagetic lesions.[54] In contrast, knockdown of TAF12 in MVNP-expressing p62P392L OCL precursors from Paget's patients decreased the 1,25-(OH)2D3 sensitivity and pagetic OCL formation, but did not affect normal OCL formation.[40] Therefore, increased TAF12 and 1,25-(OH)2D3 hypersensitivity may be necessary, but they are not sufficient to generate the full pagetic phenotype.
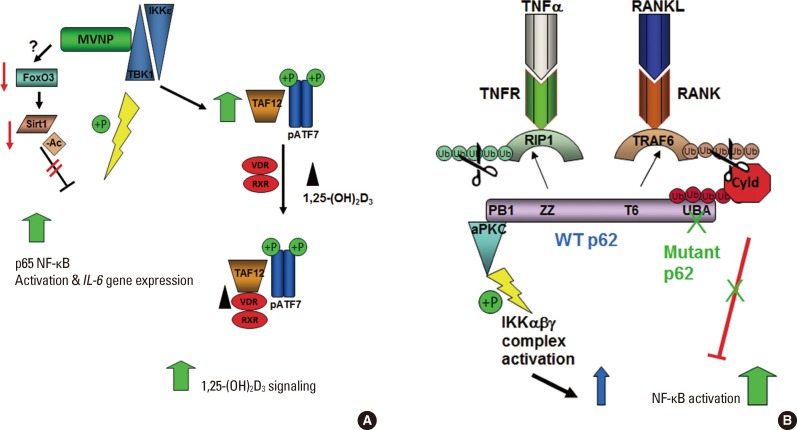
Signaling pathways altered by MVNP and mutant p62. (A) MVNP increases active TBK1, which results in increased activation of NF-κB by direct phosphorylation of S536-p65 NF-κB, leading to activation and nuclear translocation. MVNP also decreases Sirt1 deacetylation of NF-κB, leading to increased NF-κB activity. Further TBK1 is required for MVNP to increase TAF12 and phosphoATF7, leading to hypersensitivity of VDR to 1,25(OH)2D3. (B) Binding of TNF-α and RANKL to their respective receptors activates RIP1 and TRAF6, respectively, by inducing K63-linked ubiquitination. The scaffold protein p62 has specific interactions domains for both RIP1 (ZZ domain) and TRAF6 (T6 domain), an N-terminal domain that binds aPKCs (PB1 domain) and a C-terminal UBA domain that associates with ubiquitin chains. Downstream signaling via p62 pathway activation aPKC leading to activation of NF-κB is negatively regulated by recruitment of the deubiquitinase Cyld by p62 UBA interaction with the polyubiquitin chains on Cyld, and consequent TNF-α and RANKL signal attenuation by Cyld deubiquitination of RIP1 and TRAF6, respectively. Since mutant p62 does not associate with and recruit Cyld to RIP1 and TRAF6, there is less cytokine signal attenuation and, therefore, increased NF-κB activation by these cytokines. MVNP, measles virus nucleocapsid protein; TBK1, TANK-binding kinase 1; NF-κB, nuclear factor-κB; TAF12, TAF12 RNA polymerase II, TATA box binding protein (TBP)-associated factor, 20kDa; VDR, vitamin D receptor; 1,25(OH)2D3, 1,25-dihydroxy-vitamin D3; TNF-α, tumor necrosis factor-α; RANKL, receptor activator of NF-kappaB ligand; RIP1, receptor-interacting protein 1; TRAF6, TNF receptor-associated factor 6, E3 ubiquitin protein ligase; aPKCs, atypical protein kinase C; UBA, ubiquitin association; IKK, IκB kinase.
Both mutant p62 and MVNP potentiate RANKL and TNF-α responses. The mechanism by which MVNP lowers the threshold for a response to these cytokines is not well defined, although some part of the mechanism of MVNP activation of IL-6 discussed below may play a role. Both RANKL and TNF-α activate signaling pathways that involve p62, a platform protein with multiple protein-protein interaction domains that brings together upstream and downstream signaling intermediates resulting in activation of several pathways, including p38 mitogen-activated protein kinase (p38MAPK), extracellular signal-regulated kinases (ERK)1/2, and IKK, all downstream events that are important for OCL formation and function.[9] RANKL interaction with RANK results in the recruitment and activation of TNF receptor-associated factor 6, E3 ubiquitin protein ligase (TRAF6) via K63-linked ubiquitination.[55] TRAF6 then stimulates TGF-β activated kinase 1 (TAK1) binding protein (TAB)1/TAB2/TAK1-dependent[56] or atypical protein kinase C (aPKC)-dependent phosphorylation of the IKK complex,[57] leading to activation of NF-κB, as well as activating MAPK pathways to activate other key transcription factors such as AP1 and MITF. Wild type p62 has binding sites for both TRAF6 and the aPKCs (PKCζ and PKCλ) resulting in the formation of a multimeric protein complex that regulates NF-κB activation via phosphorylation of IKKβ.[57,58] TNF-α receptor TNFR1 signals, in part, via formation of a complex that includes receptor-interacting protein 1 (RIP1) binding to p62 via the p62 ZZ domain, which then leads to recruitment of aPKCs to the p62 AID domain and downstream signal activation.[59] Until recently it was a puzzle how loss function of the p62 UBA domain due to pagetic mutations led to elevated signaling. However, it was recently reported that p62 also attenuates TRAF6 and RIP1 signaling by recruiting the deubiquitinase cylindromatosis (CYLD) via binding the ubiquitin chains on CYLD (Fig. 2B).[60] CYLD deubiquitination of TRAF6, RIP1, and NF-κB-essential modulator (NEMO) leads to inactivation of NF-κB signaling. Loss of the p62 UBA function decreases p62 recruitment of CYLD to TRAF6, RIP1, and NEMO,[60] thereby decreasing the signal attenuation and leading to increased signaling at lower ligand-receptor interaction levels. Of note, optineurin, a gene in one of the other Paget's susceptibility loci can also attenuate TNF-α signaling by recruiting CYLD to RIP1.[61] Interestingly, TNF-α is more affected by loss of p62 recruitment of CYLD than RANKL, indicating that TNF-α regulation of OCL formation is more subject to negative regulation by p62 than RANKL.
Mechanisms of MVNP regulation of IL-6
Understanding the mechanism by which MVNP upregulates IL-6 expression will reveal at least part of the mechanisms by which MVNP contributes to aberrant OCL formation and increased bone formation in Paget's disease. IL-6 mRNA levels are elevated in MVNP-expressing cells. Since regulation of IL-6 mRNA levels is well known to be regulated both by transcription and post-transcription mechanisms, Wang et al.[62] analyzed IL-6 mRNA stability in the presence and absence of MVNP. They reported that MVNP increases IL-6 gene transcription, whereas IL-6 mRNA stability remains unchanged.[62] Although an array of transcription factors have been reported to regulate the IL-6 gene in response to different stimuli in a variety of cell types, all of these studies indicate that the transcription factor NF-κB is critical for basal IL-6 transcription.[63] Transcription studies have demonstrated that NF-κB is important for both basal and part of the MVNP-stimulation, indicating that there is another transcription factor that makes a significant contribution to MVNP's effects on OCL activity. In concert with this finding, MVNP has been reported to increase the levels of NF-κB activity.[41,62] The IL-6 gene is a classic example of a promoter that can be regulated by a synergistic interaction between NF-κB and the bZIP transcription factor CCAAT/enhancer binding protein (C/EBP)β.[64] Chromatin immunoprecipitation studies revealed that MVNP increases both NF-κB and C/EBPβ occupancy on the endogenous IL-6 gene promoter in NIH3T3 cells - a cell model that has replicated MVNP's induction of the IL-6 gene (unpublished data).
Studies to determine how MVNP is activating transcription factors regulating IL-6 gene expression have revealed that MVNP utilizes two pathways. MVNP was reported to be in a complex containing the IKK family members TANK-binding kinase 1 (TBK1) and IKKε and the transcription factor interferon regulatory factor 3 (IRF3).[65,66] MVNP stimulates the activation of TBK1 and IKKε, which in turn activate IRF3, and thereby IRF3 target genes, such as interferon β (INFβ). Consistent with this effect of MVNP gene expression profiling studies indicated that INFβ is upregulated in pagetic peripheral blood monocytes.[67] Activated TBK1 and IKKε can also directly phosphorylate and activate NF-κB.[68,69] Sun et al.[70] recently reported that MVNP increases TBK1 in OCL precursors via increased protein stabilization. Like expression of MVNP, TBK1 over-expression in the OCL precursor cell line RAW264.7 and in NIH3T3 cells increased activated NF-κB (phospho-S536-p65) and endogenous IL-6 mRNA, as well as increased the protein levels of TAF12 and ATF7, key players in the enhanced response of pagetic OCL precursors to 1,25-(OH)2D3. TAF12 may also have a role in the MVNP-induced increase of IL-6,[53,54] but it is unknown if it acts directly or indirectly. Lentiviral transduction of TBK1 into wild-type BM monocytes did not produce enough TBK1 protein to detect by Western blotting, so it is still unclear if elevated TBK1 is sufficient to phenocopy MVNP. However, pharmaceutical inhibition of TBK1 with BX795 impaired MVNP-induced IL-6 expression in bone marrow monocytes from TRAP-MVNP mice. Further, knockdown of TBK1 in bone marrow monocytes from TRAP-MVNP mice specifically impaired development of the MVNP-induced pagetic OCL phenotype, implying that TBK1 is required for MVNP to induce a pagetic phenotype in OCL. Therefore, TBK1 plays a critical role in mediating the effects of MVNP on IL-6 expression and OCL differentiation (Fig. 2A).[70]
The p65 subunit of the NF-κB heterodimer is subject to reversible acetylation.[71] Acetylation of p65 lysine 221 enhances DNA binding and impairs assembly with the inhibitor IκBα, while acetylation of lysine 310 is required for full transcriptional activity of p65 that is independent of changes in DNA binding or IκBα binding. The acetylation state of p65 NF-κB is controlled by the competitive influences of acetyltransferases (p300/CBP) and deacetylases (Sirtuin-1). Wang et al.[62] reported that MVNP increases NF-κB activation of the IL-6 gene by decreasing the level of Sirtuin-1 (Sirt1) expression. MVNP does this by triggering increased phosphorylation of Forkhead-box class O3 (FoxO3), which destabilizes the FoxO3 protein and leads to reduced FoxO3 induction of the sirtuin-1 gene in OCL precursors and NIH3T3 cells. Several protein kinases have been reported to down-regulate FoxO3 stability through phosphorylation, including AKT, ERK1/2, IKKβ, and IKKε.[72,73,74] Although MVNP can activate IKKε, it's not known what kinase is involved in MVNP destabilization of FoxO3 in OCL precursors. Ectopic expression of Sirt1 significantly decreased both basal and MVNP-stimulated IL-6 promoter activity. At high doses, the Sirt1 activator, resveratrol, inhibited OCL differentiation of precursors from both WT and TRAP-MVNP mice. However, a resveratrol dose that had little effect on WT OCL differentiation was able to suppress the MVNP-enhanced OCL differentiation to WT levels.[62] Thus, MVNP may act through two pathways to induce the IL-6 gene (Fig. 2A), but more investigation is needed to clarify this question.
Expression of OCL fusion molecules in OCL precursors
OCL precursors from TRAP-MVNP mice and pagetic patients expressing MVNP form OCL with increased nuclei per OCL as a result of increased cell fusion of mononuclear OCL precursors. In contrast to normal OCL, which contain 3-10 nuclei, Paget's patients OCL can contain up to 100 nuclei per OCL. The signaling that regulates OCL size and cell fusion is poorly understood. Several proteins upregulated by RANKL have been shown to have a role in the fusion process. These include dendritic cell (DC)-specific transmembrane protein (STAMP), OC-STAMP, the d2 isoform of the vacuolar ATPase V0 proton pump (Atp6v0d2), CD9, a member of the TM4 superfamily, and ADAM8.[75,76] Although both MVNP[77] and p62P392L[60] have been reported to increase the transcription factor NFATc1, which regulates OCL fusion and induces at least some of the fusion molecules (DC-STAMP and Atp6v0d2[78]), only MVNP increases OCL fusion. Teramachi and colleagues[49] measured the expression levels of several fusion molecules in TRAP-MVNP, p62P394LKI and WT OCL precursors treated with IL-6 for 4 days. OCL formed from TRAP-MVNP mice with or without IL-6 treatment had elevated expression of DC-STAMP compared with those from p62P394LKI and WT mice. The expression levels of ATP6v0d2 and ADAM8 were only modestly elevated in MVNP OCL. Little is known about the status of these fusion molecules or OC-STAMP, CD9, and the non-RANKL regulated fusion molecules CD44, CD47, and triggering receptor expressed on myeloid cells 2 (TREM2)[76] in pagetic OCL or in the presence of either MVNP or mutant p62. Thus, understanding what is perturbed in the regulation of OCL fusion that permits the hypernucleation of pagetic OCL to occur awaits further study.
Mechanism of increased bone formation induced by pagetic OCL
Bone resorption and formation are tightly linked processes, with bone formation only occurring at sites of previous bone resorption under normal bone remodeling. Paget's disease is characterized by marked increases in both OCL and osteoblast activity, and this coupled bone remodeling leads to rapid focal overproduction of poor quality bone. Further, MVNP appears to have a larger role than mutant p62 in the generating the signals leading to increased bone formation. Teramachi et al.[49] recently reported that in contrast to TRAP-MVNP mice, osteoblasts from p62P394LKI mice expressed much lower levels of Runx2 and osterix, transcription factors necessary for osteoblast differentiation, and higher levels of dickkopf WNT signaling pathway inhibitor 1 (DKK1), a Wnt antagonist. Treatment of osteoblast precursors from p62P394LKI mice with IL-6 did not increase runt-related transcription factor 2 (RUNX2) or osterix and did not decrease DKK1 levels. These results suggest that MVNP expression in OCL induces other factors in addition to IL-6, which are necessary for the development pagetic lesions in mice. The pagetic OCL play a critical role in regulating the enhanced osteoblast activity observed in Paget's patients since therapies that decrease OCL activity also decrease new bone formation and induce clinical remission in Paget's disease.[4]
Bidirectional signaling between ephrinB2 expressed on OCL and the receptor EPH receptor B4 (EphB4) expressed on osteoblasts has been implicated as a major regulator of coupling.[79,80] Increased EphB4 signaling in the osteoblast increases bone mass, whereas increased reverse signaling via ephrinB2 in the OCL suppresses OCL differentiation by inhibiting the cFos/nuclear factor of activated T-cells, cytoplasmic, calcineurin-dependent 1 (NFATc1) cascade. Recently, Hayashi et al.[81] reported that semaphorin 3A is expressed on osteoblasts and could also regulate local bone resorption and formation. They showed that semaphorin 3A binding to Nrp1 on BM monocytes suppressed OCL formation and stimulated new bone formation. Enhanced resorption releases increased growth factors from the bone matrix including insulin-like growth factor 1 (IGF1), which has a role in increasing bone matrix mineralization.[82] Teramachi et al.[2] and Kurihara et al.[83] recently reported that MVNP, but not p62P394L, increased ephrinB2, EphB4, IGF1, and semaphorin 3B in bone marrow cultures from 8-12 month old mice. Importantly, MVNP mice with IL-6 deficiency had decreased expression of ephrinB2, EphB4, and Runx2, consistent with the lack of increased bone formation in these mice. They also found that both IGF1 gene expression and protein levels were significantly enhanced in highly purified MVNP-expressing OCL. These results suggest that MVNP elevates ephrinB2/EphB4 to enhance bone formation in vitro and in vivo in TRAP-MVNP mice. Further, these data suggest that IGF1 enhances ephrinB2 expression to contribute to increased bone formation induced by MVNP (Fig. 3).
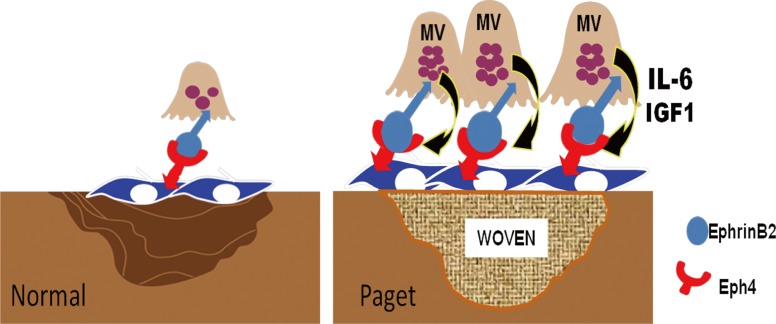
Model for the coupling of bone resorption to bone formation in Paget's disease. MVNP expression in pagetic OCL results in high levels of IL-6 that induce ephrinB2 on OCL and EphB4 on osteoblasts to increase coupled bone formation. MVNP also induces IGF1 expression by OCL that further drives bone formation and increases ephrinB2 on OCL. MVNP, measles virus nucleocapsid protein; OCL, osteoclasts; IL-6, interleukin-6; EphB4, EPH receptor B4; IGF1, insulin-like growth factor 1.
Other bone formation factors that are elevated in Paget's include fibroblast growth factor 2 (FGF2), which was reported to be elevated in the serum of Paget's patients and increased by MVNP expression in OCL precursors.[84] Interestingly, FGF2 was reported to increase osteoblast expression of IGF1,[85] which may work together with the OCL-produced IGF1 to enhance bone formation. This group also recently reported that both the chemokine (C-X-C motif) ligand 5 (CXCL5) mRNA was also increased both in sera from Paget's patients and by MVNP expression in OCL precursors.[86] Both FGF2 and CXCL5 increase stromal cell production of RANKL, thereby enhancing OCL formation in a feedback loop. In addition, Teramachi et al.[49] reported that marrow stromal cells from p62P394LKI/TIL-6 mice expressed higher levels of RANKL in response to 1,25-(OH)2D3 than the marrow stromal cells from the other mouse genotypes (TRAP-MVNP, TIL-6, p62P394LKI, WT). The stromal cells from p62P394LKI/TIL-6 also expressed high levels of TAF12. The expression of TAF12 in stromal cells can result in hyper-responsivity to 1,25-(OH)2D3. These findings may in part explain the enhanced RANKL production present in the marrow microenvironment of pagetic patients.
Future directions
Paget's disease has provided important insights into normal bone remodeling and OCL regulation of new bone formation. It will be important to determine the mechanisms responsible for the exuberant bone formation in Paget's disease, as a means to identify potential new anabolic agents for bone that might be active in patients with osteoporosis. Further, understanding the mechanisms of abnormal bone remodeling in Paget's disease should provide important insights into the pathophysiology of inflammatory bone diseases and bone metastases, and provide novel therapeutic targets for these diseases.
Notes
No potential conflict of interest relevant to this article was reported.
GDR is a consultant to Amgen. DLG states that she has no conflicts of interest.