Two Types of Mouse Models for Sarcopenia Research: Senescence Acceleration and Genetic Modification Models
Article information

Abstract
Sarcopenia leads to loss of skeletal muscle mass, quality, and strength due to aging; it was recently given a disease code (International Classification of Diseases, Tenth Revision, Clinical Modification, M62.84). As a result, in recent years, sarcopenia-related research has increased. In addition, various studies seeking to prevent and treat sarcopenia by identifying the various mechanisms related to the reduction of skeletal muscle properties have been conducted. Previous studies have identified muscle synthesis and breakdown; investigating them has generated evidence for preventing and treating sarcopenia. Mouse models are still the most useful ones for determining mechanisms underlying sarcopenia through correlations and interventions involving specific genes and their phenotypes. Mouse models used to study sarcopenia often induce muscle atrophy by hindlimb unloading, denervation, or immobilization. Though it is less frequently used, the senescence-accelerated mouse can also be useful for sarcopenia research. Herein, we discuss cases where senescence-accelerated and genetically engineered mouse models were used in sarcopenia research and different perspectives to use them.
INTRODUCTION
Research on sarcopenia has become more active in recent years as prevalence has risen worldwide.[1] Meanwhile, the International Classification of Diseases, Tenth Revision, Clinical Modification (ICD-10-CM) of the USA, based on the ICD-10, a global classification of diseases published by the World Health Organization, issued the disease code (M62.84) to sarcopenia in 2016.[2]
With the proper disease classification now on the books, sarcopenia has become the focus of many medical scientists actively conducting studies to find treatments. In vivo murine experiments are perhaps the most useful methods for investigating effective medications or interventions for sarcopenia because they have the advantage of being able to analyze the effects of interventions both on genotype and on phenotype.[3]
In general, methods of inducing muscular atrophy in mice by hindlimb unloading, denervation, or immobilization are widely used to study sarcopenia, though there are also methods used aged-mice.[3] Compared to the mouse models mentioned earlier, the range of applications for accelerated senescence and genetically modified mouse models in research may be relatively narrow, but there are mouse models that can be reasonably used in sarcopenia research according to the purpose of the study.
The accelerated senescence mouse has the advantage that it can dramatically shorten the experimental period compared to experiments using aged mice. As far as genetically modified mouse models have the advantage of being able to determine whether a specific medication or intervention is able to stimulate or improve a specific pathway associated with sarcopenia. In this review, we introduce accelerated senescence mouse models and genetically modified mouse models that can be used in sarcopenia research (Fig. 1).
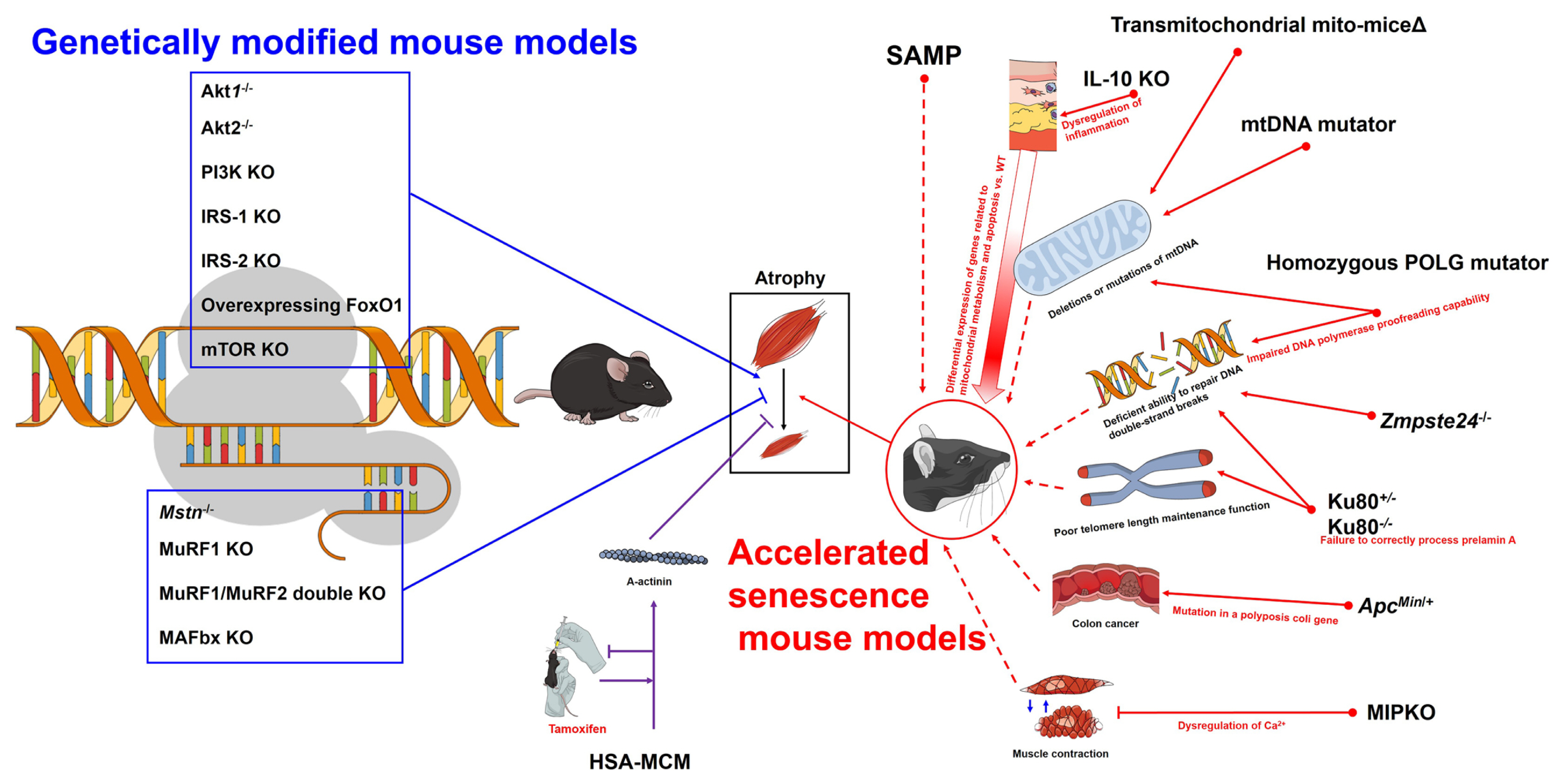
Overview of accelerated senescence mouse models and genetically modified mouse models for sarcopenia research. Akt, protein kinase B; PI3K, phosphoinositide 3-kinase; KO, knock-out; IRS, insulin receptor substrate; FoxO, forkhead box O; mTOR, mammalian target of rapamycin; Mstn, myostatin; MuRF, muscle ring finger; HSA-MCM, human skeletal muscle actin Mer-Cre-Mer; SAMP, senescence-prone inbred strains; WT, wild-type; IL, interleukin; mtDNA, mitochondrial DNA; POLG, DNA polymerase γ; Zmpste24, zinc metallopeptidase STE24; ApcMin/+, adenomatous polyposis coli multiple intestinal neoplasia; MIPKO, muscle-specific inositide phosphatase knock-out.
ACCELERATED SENESCENCE MOUSE MODELS
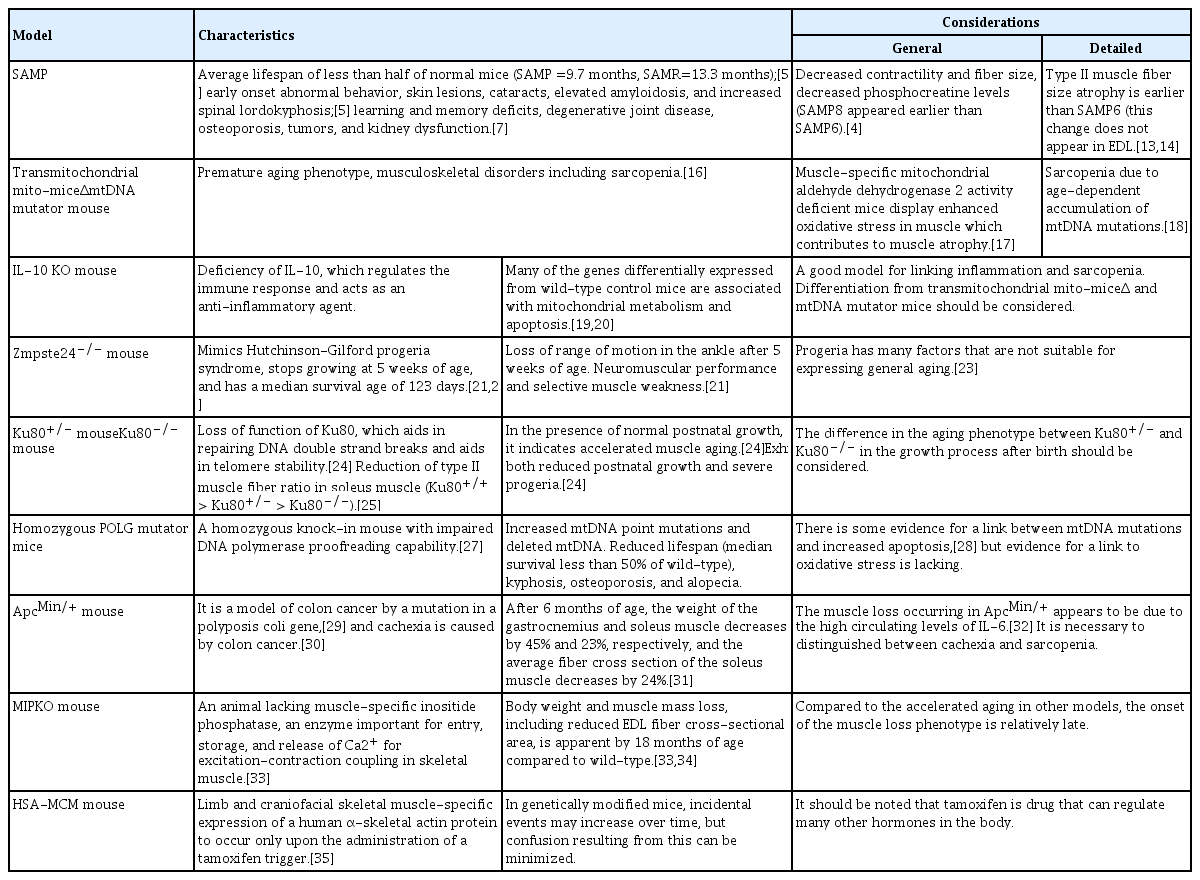
As mentioned in our previous paper, natural aged mice are the best models for studying sarcopenia because they have similar morbidities to those of human sarcopenia patients. Unfortunately, these models are limited by high cost and low availability of experimental animals.[3] Thus, several mouse models with accelerated aging phenotypes have been developed, enabling an accelerated observation of aging’s influence on muscle physiology and form. Accelerated models of aging afford us the convenience of examining the effects of aging earlier in the life of the animal, shortening the requisite wait for senescence. However, accelerated aging models do not always demonstrate characteristics typical of aging for the animal being studied (Table 1).
Accelerated senescence mouse models for sarcopenia research
One of the most commonly used accelerated aging models is the senescence-accelerated mouse (SAM).[4] SAM has been under development at Kyoto University since 1970 and was created by selective sister-brother mating of AKR/J mice donated by the Jackson Laboratory (Bar Harbor, ME, USA) in 1968.[5,6] Lineages of this mouse include 14 senescence-prone inbred strains (SAMP) and 4 senescence-resistant inbred strains (SAMR), with mean life spans of 9.7 and 13.3 months, respectively.[5] Characteristic aging features in the senescence-prone mice include abnormal behavior, skin lesions, cataracts, elevated amyloidosis, and increased spinal lordokyphosis.[5] Other pathologies simulating aging, such as learning and memory deficits, degenerative joint disease, osteoporosis, tumors, and kidney dysfunction, have also been identified.[7] Research to determine whether SAMP6 [8] and 8 [9] were appropriate muscle aging models investigated fiber size and quadriceps tubular aggregate accumulation, respectively. Derave and co-workers [4] examined several properties of muscle physiology, including muscle metabolites, muscle mass, contractile properties, force generation, fatigability, fiber size, and distribution among 2 strains of senescent-accelerated (SAMP6, SAMP8) and 1 strain of SAMR1 mice. All mice were studied at 10, 25, and 60 weeks of age.[4] Both accelerated aging models displayed reduced muscle contractility, decreased fiber size, and increased muscle phosphocreatine levels with aging. However, SAMP8 exhibited the features most demonstrative of accelerated muscle aging with a greater reduction in muscle phosphocreatine levels, muscle mass, contractility, and type II fiber size, with aging features developing earlier and at a faster rate than SAMP6 or SAMR1. Muscle fiber composition in the soleus showed a shift toward more type I fibers over time in both SAMP6 and SAMP8; however, the difference was not significant. No fiber size or type distribution change was significant in the extensor digitorum longus (EDL) of either SAMP6 or SAMP8. These findings suggest that the SAMP8 mouse soleus is a reasonable model for muscular aging studies.
SAM, which consists of 18 lines (11 SAMP and 7 SAMR), are regarded as a good choice for the study of sarcopenia.[6,10] According to a previous study on SAMP8 mice at 10 (young), 25 (adult) and 60 (old) weeks of age,[4] SAMP8 mice exhibited the typical features of accelerated muscle aging with a short life span and rapid aging due to high oxidative stress,[11,12] a greater decrease in muscle mass and contractility, and a larger reduction in the size of type II muscle fibers than wild-type (WT) animals.[13,14] All findings indicated that the SAMP8 mouse model is a reasonable model for sarcopenia research. However, to date, there are no reports investigating when sarcopenia occurs in the SAMP8 model. The onset in the alteration of muscle mass and function, as well as muscle structural alteration, should be investigated in the SAMP8 model in future research. SAMP8 showed peak muscle mass at month 7 and the onset of an ex vivo contractility decline of the gastrocnemius was observed from month 8 onwards. Compared with month 8, most of the functional parameters measured decreased significantly at month 10 in SAMP8 mice.[15]
The transmitochondrial mito-miceΔ and mitochondrial DNA (mtDNA) mutator mice display premature aging phenotypes and display a spectra of musculoskeletal disorders, including sarcopenia.[16] In muscle-specific mitochondrial aldehyde dehydrogenase 2 activity-deficient mice, enhanced oxidative stress in muscle contributes to muscle atrophy.[17] The age-dependent accumulation of mitochondrial DNA mutations leading to mitochondrial dysfunction is considered to be an important contributor to sarcopenia.[18] However, microarray analysis of skeletal muscle gene expression in the frail mouse interleukin (IL)-10 knock-out (KO) model showed many of the 125 genes differentially expressed between 50-week-old IL-10 KO and WT C57BL/6 litters were associated with mitochondrial metabolism and apoptosis.[19,20] Therefore, it is up to the investigator to decide whether to select the transmitochondrial mito-miceΔ and mtDNA mutator mice or the IL-10 KO mice for sarcopenia research, and the researcher should make that decision based on their proposed hypothesis and area of focus.
The genetic manipulation performed in developing these animals often affects the physiology of other systems, influencing results in the tissues studied. The zinc metallopeptidase STE24 (Zmpste24)−/− mouse was designed to mimic Hutchinson-Gilford progeria syndrome and stops growing at 5 weeks of age with a median survival age of 123 days.[21,22] Zmpste24−/− mice display reduced grip strength and whole-body tension when controlled for body size. In previous work, the Zmpste24 KO model demonstrated progressive ankle range of motion losses after 5 weeks of age while the WT retained its ankle mobility. Additionally, passive ankle torques in Zmpste24−/− mice, indicating increased joint stiffness.[21] The general phenotype is one of impaired neuromuscular performance and selective muscle weakness when compared to the Zmpste24+/+ littermates, which are characteristics matching those of aged mice. How well this model represents accelerated aging is controversial, as humans with progeria exhibit some aspects of rapid aging but not others.[23] The Ku80+/− (Ku80 is a protein encoded by the Xrcc5 gene) mouse is an animal that displays accelerated muscle aging with otherwise normal postnatal growth. The Ku80 KO mouse exhibits both reduced postnatal growth and severe progeria. The Ku80 heterodimer assists in the repair of DNA double-strand breaks and helps with telomere stability.[24] Didier and colleagues [25] found that the Ku80−/− mice displayed an increased proportion of slow fibers with a decreased proportion of fast fibers in soleus muscle compared to young Ku80+/+ mice. Ku80+/− animals also demonstrated a size reduction in all soleus fiber types compared to Ku80+/+. The Ku80−/− mouse is smaller than the Ku80+/+ mouse [26] and smaller type I and II fibers were found in the KO mouse.[25] Another model of accelerated aging which mimics sarcopenia is the DNA polymerase γ (POLG) mouse, a homozygous knock-in mouse with impaired proofreading capability of its mitochondrial DNA polymerase.[27] This mouse exhibits increased mtDNA point mutations and deleted mtDNA. Its reduced life span (median survival less than 50% of WT) and physical traits such as spinal kyphosis, osteoporosis, and alopecia suggest a causal relationship between the mtDNA mutations and premature aging. Although they found no evidence of elevated oxidative stress in the presence of these mutations, Kujoth and colleagues [28] discovered an increased induction of apoptosis by examining caspase-3 activation in several tissues at earlier ages. Another mouse model developed in 2008 exhibits the frailty seen in the elderly.[20] This “frail” mouse is characterized by a homozygous deletion of the Il10 gene, an anti-inflammatory cytokine (IL-10) coding gene. The absence of this IL promotes the expression of nuclear factor (NF)-κB inflammatory mediators. To mimic human frailty, typical characteristics of muscle weakness, inflammation, decreased physical function, and overall reduced activity should appear later in life. However, the frail mouse model has a disadvantage in that it only shows the weakness of the body in which independent genes and proteins are involved, rather than imitating human frailty.
The adenomatous polyposis coli multiple intestinal neoplasia (ApcMin/+) mouse is a genetic model of colon cancer that is triggered by a mutation of the polyposis coli gene,[29] leading to intestinal polyps and eventually tumor formation.[30] This mouse has also become a model of cachexia, with a significant loss of muscle and fat tissue by 6 months of age.[30,31] At 3 months of age ApcMin/+ and C57BL/6 mice have similar body weights and gastrocnemius and soleus sizes; however, by 6 months, gastrocnemius and soleus muscle weights fall by 45% and 25%, respectively, with the soleus muscle mean fiber cross-sectional area decreasing by 24%. IL-6 is a cytokine known for its cachectic impact, and the ApcMin/+ mouse exhibits elevated levels of circulating IL-6 compared to age-matched WT controls. An ApcMin/+/IL-6−/− (IL-6 null) was created which showed muscle mass similar to controls at 26 weeks of age, as opposed to the ApcMin/+ strain where a significant loss of gastrocnemius weight was detected by the same age.[32] Additionally, gastrocnemius mass was reduced by 23% with IL-6 overexpression and 32% in ApcMin/+ and ApcMin/+/ IL-6−/− mice.[32] The muscle-specific inositide phosphatase KO (MIPKO) mouse is an animal lacking MIP, an enzyme important for entry, storage, and release of the Ca2+ required for excitation–contraction coupling in skeletal muscle.[33] This animal demonstrates several characteristics consistent with premature aging relative to muscle structure and function.[34] For instance, the MIPKO is spontaneously less active and reaches exhaustion earlier than similarly aged WT littermates. Body weight and muscle mass loss, including reduced EDL fiber cross-sectional area, is apparent by 18 months of age in MIPKO animals compared to WT controls.[33,34]
The models listed above are all examples of the constitutive influences of genetic expression on biochemical pathways. As these animals assume their KO or transgenic (Tg) genotype, they will experience that influence from the time of conception, through development, and to the end of life. Sarcopenia is a post-maturational phenomenon whose process could be altered and therefore be inaccurately represented when influenced by these constitutive models. An inducible model exists in which muscle gene expression can be initiated after attaining maturity.[35] The human skeletal muscle actin Mer-Cre-Mer (HSA-MCM) mouse allows for limb and craniofacial skeletal muscle-specific expression of a human α-skeletal actin protein to occur only upon the administration of a tamoxifen trigger.[35] This type of model enables researchers to assess protein expression at any point during the animal’s life span, minimizing the confounding influences that genetic manipulation might elicit when spanning great periods or crossing developmental transition points.
As described above, several mouse models with an accelerated senescence phenotype can shorten the duration of studies to observe the effect of aging on muscle physiology and morphology (e.g., sarcopenia). However, since accelerated senescence mouse models do not always show typical characteristics of aging in the animals under study, it is important to clearly understand the pathways or causes that accelerated senescence (Fig. 1).
GENETICALLY MODIFIED MOUSE MODELS
Numerous pathways are involved in the biochemical and physiological processes of sarcopenia, so genetic mutant mouse models have been created to relate these pathways (Table 2). In sarcopenia, these pathways display an imbalance between anabolism and catabolism, with protein loss ultimately dominant and a stronger influence than protein gain.[36] Endocrine and inflammatory factors are generally mediated in this process.[37] Catabolic pathways involved in the loss of skeletal muscle mass that could play a role in sarcopenia include the endosome–lysosome, Ca2+-dependent, caspase-dependent, and ubiquitin/proteasome-dependent pathways.[38] Growth hormone, insulin-like growth factor 1 (IGF-1), insulin, and testosterone, along with adequate nutrition and exercise, play roles as positive regulators of skeletal muscle synthesis.[36,39,40] The first central pathway that controls muscle size is the phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) pathway, modulated by IGF-1 and insulin.[41] IGF-1 and insulin are involved in the stimulation of protein synthesis and hypertrophy. Interaction with their respective tyrosine kinase receptors causes phosphorylation of insulin receptor substrate (IRS)-1 which activates PI3K/Akt signaling, thereby activating the mammalian target of rapamycin (mTOR), and phosphorylating the target ribosomal p70 S6 kinase (p70S6K) and 4E-binding protein 1 (4E-BP1).[42–45] Activation of this pathway results in resistance to atrophy by phosphorylation of forkhead box O (FoxO) transcription factors, preventing their translocation to the nucleus where they would otherwise promote transcription of atrophy-related genes, including ubiquitin ligase, that goes on to degrade muscle protein.[46,47]
Genetically modified mouse models for sarcopenia research
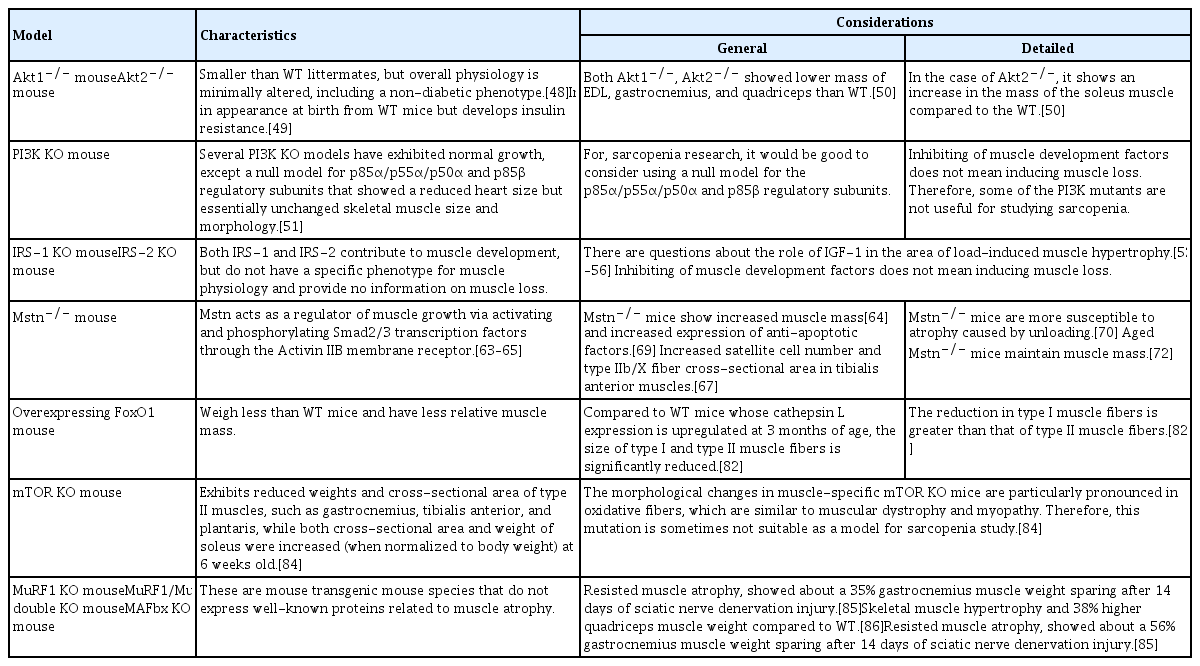
The Akt1−/− (akt 1 null) mouse is smaller than WT littermates, but the overall physiology is minimally altered with a non-diabetic phenotype.[48] On the other hand, the akt2 null mouse is indistinguishable in appearance at birth from WT mice, but develops insulin resistance.[49] Akt1−/− decreased the mass of EDL, gastrocnemius, and quadriceps compared to WT. Akt2−/− also demonstrated a decrease in EDL and gastrocnemius mass but showed an increase in mass in the soleus when compared to WT animals. The mean cross-sectional area of glycolytic EDL was reduced in both Akt1 null and Akt2 null mice compared to WT. Interestingly, the oxidative soleus muscle cross-sectional area was not different between genotypes.[50]
Several PI3K KO mouse models exhibit normal growth, except the null models for the p85α/p55α/p50α and p85β regulatory subunits that display a reduced heart size but essentially unchanged skeletal muscle size and morphology.[51] The downstream effects of insulin and IGF-1, IRS-1, and IRS-2 KO show no specific changes to the phenotype of muscle physiology. Both IRS-1 and IRS-2 are activated by insulin, growth hormone, and IGF-1, and contribute to muscle development but do not seem to have any input on muscle loss. So, some of these PI3K mutants and the IRS-1 and IRS-2 KO mice may not be as useful for studying sarcopenia.
The role of IGF-1 in the area of load-induced muscle hypertrophy has been questioned recently. Mechanical loading can activate mTOR complex 1 signaling and promote muscle hypertrophy.[52–54] Also, the hypertrophic response induced by mechanical overload is fully preserved in muscles from Tg mice that express a dominant-negative IGF-1 receptor (IGF-1R) specifically in skeletal muscle.[55] Furthermore, IGF-dependent signaling toward enhanced protein synthesis via the Akt-mTOR-S6K pathway is not always observed after strength type exercise in humans or in mice. Tg mice expressing mutant IGF-1R in skeletal muscle (MKR) stimulate IGF-1R despite the absence of functional IGF-1.[55,56] The MKR mice expressed high Akt and p70S6K due to continuous stimulation of IGF-1.[55,56] In contrast to KO mice, Akt Tg mice overexpressed the constitutively active form of Akt and showed marked hypertrophy of myotubes, increased muscle atrophy resistance, and maintenance of fiber size distribution following denervation.[41,45]
NF-κB is a transcription factor that activates the expression of muscle ring finger 1 (MuRF1; a protein encoded by the Trim63 gene) by binding to its promoter and initiates proteasome-induced muscle degradation.[57] NF-κB is highly expressed in disuse atrophy and cachectic conditions, but not typically in sarcopenia.[38,58–62]
Myostatin (gene name Mstn) acts as a negative regulator of muscle growth via activating and phosphorylating Smad2/3 transcription factors through the Activin IIB membrane receptor.[63–65] The myostatin and IGF-1/PI3K/Akt pathway are interact via a negative feedback loop.[66] Myostatin levels increase during disuse atrophy and cachexia, and inhibition of myostatin induced muscle hypertrophy (increase muscle fiber size) and muscle mass gain.[67,68] Myostatin KO mice (Mstn−/−) show increased muscle mass [64] and increased expression of anti-apoptotic factors.[69] Also, Mstn−/− mice display increased satellite cell numbers and type IIb/X fiber cross-sectional area in tibialis anterior muscles.[67] Interestingly, a recent study reported that Mstn−/− mice are more susceptible to atrophy from unloading.[70] Although myostatin’s role in sarcopenia is controversial and even though the expression of Mstn mRNA ranged from decreased, to not changed, to increased in aging rodent models,[71] the aged Mstn−/−mice maintained their muscle mass.[72] These results indicate that myostatin acts as a negative regulator of muscle mass in older subjects and suggests that the Mstn−/− mouse could be a useful model for studying age-related muscle loss.[72]
Autophagy is a catabolic cellular process for clearing and recycling dysfunctional organelles and denatured proteins in response to nutrient deprivation, viral infection, and genotoxic stress.[73,74] Autophagy can promote either cell survival or cell death depending on the circumstance, and both excessive and defective autophagy is highly correlated with the loss of skeletal muscle.[75] Various intrinsic and extrinsic factors are involved in autophagy. Oxidative stress or extreme catabolic conditions can up-regulate autophagy.[76–79] Although mTOR is a key regulator of autophagy pathways,[80] FoxO3 is the main inducer of autophagy genes such as LC3 (mouse gene name Map1lc3a) and Bnip3 in skeletal muscles.[81]
Tg mice overexpressing FoxO1 weighed less than WT mice and had a lower relative muscle mass. 3-month-old FoxO1 Tg mice exhibit a marked decrease in the size of type I and II fibers and a significant decrease in the number of type I fibers compared to WT mice. FoxO1 Tg mice induce an increase in cathepsin L, a lysosomal proteinase, along with muscle atrophy.[82] Similar to Tg mice, FoxO1 null mice also exhibit a reduction in slow-twitch fiber formation in the soleus muscle but unchanged type II fiber plantaris muscle composition.[83]
Muscle-specific mTOR KO mice exhibit reduced weights and cross-sectional areas of type II muscles, such as gastrocnemius, tibialis anterior, and plantaris muscles, while both the cross-sectional area and weight of the soleus was increased (when normalized to body weight) at 6 weeks old.[84] The morphological changes of muscle specific mTOR KO mice resembles the myopathy of muscular dystrophy due to especially prominent oxidative fibers; this mutant may not serve well as a sarcopenia model.[84]
MuRF1 and MAFbx (also known as Atrogin1 or F-box protein 32, a protein encoded by the Fbxo32 gene) are well-known proteins involved in muscle atrophy. KO for MuRF1 or MAFbx conferred resistance to muscle atrophy, with about 36% and 56% gastrocnemius muscle weight spared after 14 days of sciatic denervation injury, respectively.[85] MuRF1/MuRF2 (a protein encoded by the Trim55 gene) double-KO mice also showed skeletal muscle hypertrophy and a 38% increased quadriceps weight compared to WT animals.[86]
The translation of various genes into proteins and their actions play an important role in muscle synthesis and degradation. The genetically modified models can be important in that it can be applied to research by understanding and approaching the physiological changes in muscle at the molecular level. These genetically modified models could help understand the effects of specific interventions (drugs, exercise etc.) on specific cellular signaling pathways.
POINTS TO CONSIDER WHEN SELECTING A MOUSE MODEL FOR SARCOPENIA RESEARCH
As mentioned in our previous paper, there are differences in overall lifespan and muscle fiber types, and thus differences in energy metabolism, between mice and humans.[3] Therefore, to properly choose a mouse model for sarcopenia research, it is essential to recognize these differences.
As described earlier, in the case of accelerated senescence mice, various pathologies appear simultaneously with rapid aging.[5,7] Therefore, when evaluating the mass and quality of skeletal muscle in accelerated senescence mice, it will be necessary to identify all pathologies that appear at the time of testing. Among the accelerated senescence mice models, both SAMP6 and SAMP8 show rapid aging and decreased skeletal muscle mass.[8,17] In particular, in the case of SAMP8, the decrease in phosphocreatine levels, skeletal muscle mass, and type II muscle fibers appeared earlier and faster than SAMP6 or SAMR1.[4] In other skeletal muscle regions of SAMP8, the size of the muscle bundle and the ratio of the types of muscle fiber do not change. On the other hand, in the soleus muscle of SAMP8 mice, the ratio of muscle fiber type changes, decreasing type II fibers while increasing type I fibers during rapid aging [4]; this change in fiber composition occurs prominently between 8 and 10 months of age. Taken together, these data suggest that 8 to 10-month-old SAMP8 mice may be ideal for studying sarcopenia.
The transmitochondrial mito-miceΔ and mtDNA mutator mice may be used accurately to study sarcopenia with the assumption that sarcopenia is caused mitochondrial dysfunction.[16] However, since IL-10 KO mice were also found to display increased mitochondrial metabolism and apoptosis,[19,20] it would be up to the investigator whether to select these models for their intended experiments. In addition, there are various other accelerated senescence mouse models such as Zmpste−/−, Ku80−/−, ApcMin−/−, MIPKO that can be used in sarcopenia research. To use these models properly, a model should only be selected after a sufficient understanding of the different senescence acceleration mechanisms has been obtained. Without this required understanding, experimental design becomes near impossible and results uninterpretable.
Genes encoding proteins related to muscle synthesis or differentiation inhibition can be engineered and used in sarcopenia studies. Genetically modified mice may be suitable for evaluating medications that inhibit or stimulate specific biological pathways, and it is believed that the selection of a model that fits the target pathway is vital. The mouse models mentioned in this study should be selected and used after grasping the genetic homogeneity with humans and a deep understanding of the mechanisms related to aging, muscle synthesis, and muscle differentiation inhibition.
Notes
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MEST) (No. NRF-2019R1F1A1059208).
Ethics approval and consent to participate
Not applicable.
Authors’ contributions
Conceptualization: KWB, YKJ, and JIY; Investigation: JSP, JSK, and YSH; Writing–original draft preparation: KWB, YKJ, and SJK; Writing–review and editing: KWB, YKJ, JSP, JSK, YSH, SJK, and JIY; Project administration: JIY; Funding acquisition: JIY; All authors read and approved the final manuscript.
Conflict of interest
No potential conflict of interest relevant to this article was reported.