INTRODUCTION
Hormone resistance is a condition characterized by reduced organ responsiveness to a hormone, as a result of either defective hormonal receptor or impaired post-receptor binding signaling. In 1942, Albright et al. [1] was the first to report a hormonal resistance syndrome, by describing a disease that shares the same biochemical characteristics with hypoparathyroidism in the context of elevated parathyroid hormone (PTH) levels. Pseudohypoparathyroidism (PHP) and related disorders are rare and heterogenous diseases, sharing molecular defects in the PTH-related protein (PTHrP) signaling pathway with variable clinical presentation and severity.[2] The aetiology of PHP and related disorders resides in guanine nucleotide-binding protein, α-stimulating (GNAS) gene, a locus that encodes the α-subunit of guanine nucleotide-binding G-protein α-subunit (Gsα), via an imprinting phenomenon.[2]
The first global consensus on the disease, released in 2018, recognizes PHP as a primarily clinical diagnosis.[2] The diagnostic procedure should be based on the presence of hormonal resistance to PTH and/or early-onset obesity and/or Albright’s hereditary osteodystrophy (AHO) phenotype -consisting in major of brachydactyly type E and short stature and minor of stocky build, round face and ectopic ossifications.[2] Short stature and brachydactyly type E of the AHO phenotype are possibly due to a Gsα haploinsufficiency in bone tissue, independent of the affected allele parental origin, while the hormonal resistance to PTH-biochemically expressed as hypocalcemia, hyperphosphatemia - is apparent only when mutations are located on the maternally inherited allele.[3] Thus, PHP type 1A (PHP1A) is diagnosed when the affected GNAS allele is maternally originated, whereas when the mutated haplotype is of paternal origin, a possible diagnosis is pseudo-PHP or progressive osseous heteroplasia. Since PTH-PTHrP signaling pathway defects present overlapping clinical signs, the gold standard procedure for the assignment of the PHP subtype diagnosis is molecular genetic testing.[2]
In this case report, we present a 12-year-old boy with a novel de novo GNAS mutation in the maternally transmitted allele, with a mild atypical skeletal phenotype, diagnosed in a totally healthy family of Greek origin. Approval of the index patient and consent of parents was given for the report of this case.
CASE REPORT
A 12-year-old boy was presented at the outpatient clinic of the Unit of Paediatric Endocrinology and Metabolism, for evaluation of short stature and unusual finger phenotype.
He was the first offspring of two phenotypically healthy non-consanguineous parents with no obvious skeletal deformities, and had three phenotypically healthy sisters, normal growth and no chronic disease. In the family, the paternal grandfather was diagnosed with osteoporosis at the age of 60, the maternal grandmother with hypothyroidism, and the maternal grandfather with type 2 diabetes mellitus.
The index-patient was born after a full-term pregnancy after spontaneous conception. At 12 weeks of gestation, vaginal bleeding due to a mild placental abruption was dealt with a 21-day bed rest. At birth, the newborn exhibited a normal adaptation to extra-uterine life, with z-score values of −2.84 for birth weight, −1 for length and −2.73 for head circumference. Developmental and motor milestones were achieved at normal age.
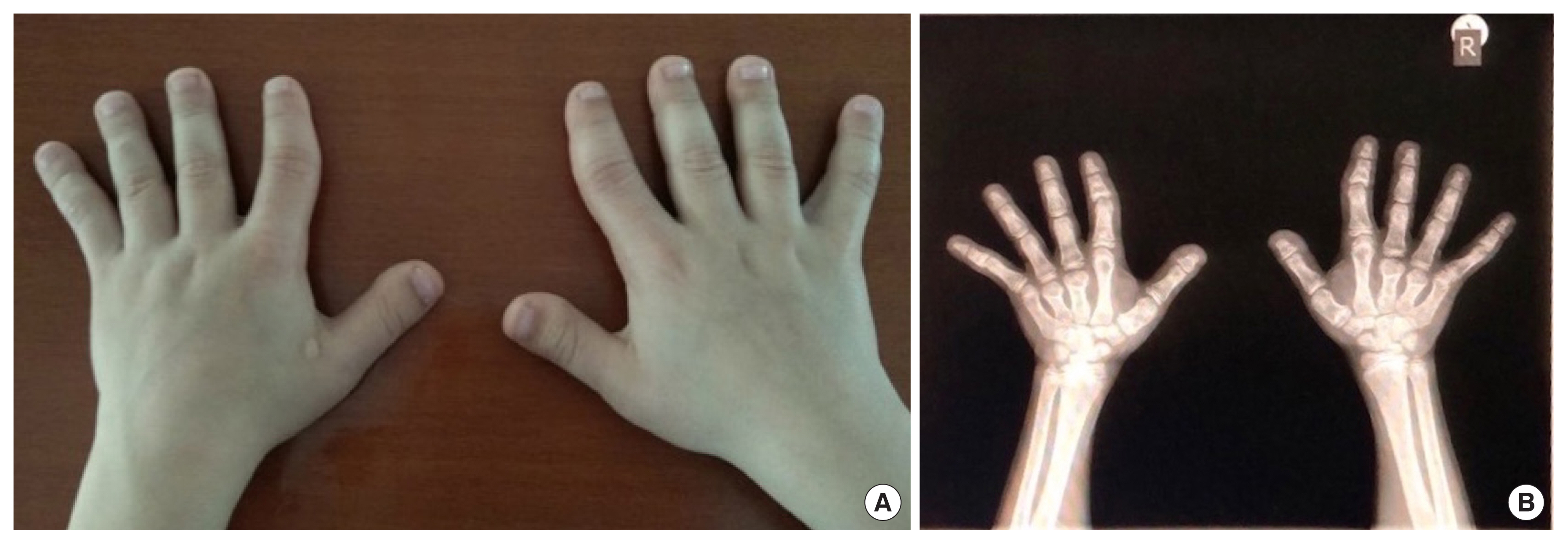
At the age of 12 years, anthropometric measurements revealed a symmetrical short and lean boy with a height z-score of −1.58 (1.37 m, 6th percentile), a weight z-score of −2.49 (27,000 g, <3rd percentile), a head circumference z-score of −1.28 (0.522 m, 10th percentile) and low body mass index (14.4 kg/m2, z-score, −2). Tanner pubertal stage was recorded as stage I, but volume of testes (4 mL) suggested the clinical beginning of puberty. Physical examination identified signs of AHO, including a round face, a depressed nasal bridge, a short neck, micrognathism, arched palate, spots on the skin of the face, brachydactyly of all digits of the hands and feet as well as clinodactyly (Fig. 1A). No subcutaneous ossification was observed.
The hand X-ray revealed short, broad and dysmorphic metacarpal bones of the 3rd, 4th and 5th digit of both hands along with the absence of secondary ossification centers of the 3rd and 4th metacarpals and the proximal interphalangeal joint of the 2nd and 5th digits of the hands (Fig. 1B). The estimated bone age according to Greulich and Pyle was 11 6/12 years, compatible with chronological age. The cranial findings of magnetic resonance imaging were normal.
Our first laboratory examination revealed normal total serum calcium levels, normal ionized calcium levels, and relatively low phosphorus levels, with a mildly elevated serum PTH level (Table 1). Anti-thyroid peroxidase and thyroglobulin autoantibodies were negative. Laboratory investigation of failure to thrive investigating haemoglobin, total cholesterol, folic acid, B12, ferritin levels was normal, and in addition, celiac disease was excluded.
Genetic assessment of the proband and his parents was performed in genomic DNA and direct sequencing identified a substitution of guanine for adenine at nucleotide 389 (c.389 A>G) in exon 5, leading to an amino acid replacement in codon 130 of tyrosine by cysteine (p.Tyr130Cys). Analysis of GNAS loci of both parental genomic DNA failed to identify any base substitution in the analyzed region. Analysis of the origin of the proband’ s mutated allele revealed the maternal origin of the de novo c.389 A>G GNAS haplotype.
To the best of our knowledge, this is a novel previously unreported mutation of GNAS in the Human Gene Mutation Database (http://www.HGMD.cf.ac.uk/ac/index.php). The fact that only the affected proband was a carrier of this novel mutated allele implies that the detected sequence alteration is a disease-causing mutation. According to the Human Genome Variation Society nomenclature of the recommendations of the American College of Medical Genetics and Genomics-Association for Molecular Pathology, the hereby reported mutation c.389 A>G (p.Tyr130Cys) of the exon 5 in GNAS gene, is classified as likely pathogenic (class 4).[4] The identification of the mutation, in liaison with the phenotype and the index laboratory profile, confirmed the presumptive diagnosis of a PHP1A (inactivating PTH/PTHrP signaling disorder 2 [iPPSD2]).
DISCUSSION
PHP1A is a clinical entity mainly caused by maternally transmitted heterozygous inactivating molecular defects of the GNAS gene, leading to a defective signaling pathway of G protein-coupled receptors. According to the EuroPHP network nomenclature, the disease is also discussed in the literature as iPPSD2.[5] Affected individuals present either resistance to a single hormone -mainly PTH- or to a combination of hormones such as PTH, thyroid stimulating hormone, follicle-stimulating hormone, luteinizing hormone, or growth hormone-releasing hormone.[2] The expression of the GNAS gene in the target tissue of the above-mentioned hormones is subjected to imprinting.[3] In this case report, we present a patient with a mild PHP1A course with a specific isolated bone phenotype including brachydactyly and relatively short stature, manifesting normocalcaemia due to mild PTH resistance and no resistance to other hormones, until the age of 12 years. The molecular defect leading to the phenotype is the previously unreported c.389 A>G point mutation in exon 5 of GNAS gene that leads to the replacement of p.Tyr130Cys of Gsα peptidic strain.
In tissues with biallelic GNAS expression, molecular defects in heterozygous state do not compromise gene function.[3] In growth plate chondrocytes however, heterozygous mutations can interfere with adequate gene expression and normal plate development. Currently, more than 400 frame-shift, missense, nonsense, splice-site, and partial or whole deletion or insertion mutations of the GNAS gene lead to an altered Gsα protein of compromised functionality, with no absolute genotype-phenotype correlation.
PTH-related peptide receptor (PTH1R) and PTH are able to modify the differentiation of chondrocytes.[6] More precisely, PTH1R receptor apart from activating Gs and thus, the adenylyl cyclase pathway, also activates another heterotrimeric G protein subunit, the Gq/11 subunit that is responsible for the activation of the phospholipase C pathway.[6] Thus, activation of PTH1R may be a critical regulator of bone maturation. It has been already reported that loss of Gsα function causes chondrocyte hypertrophy, whereas loss of Gq function leads to delayed hypertrophy, in murine growth plates.[6] Based on these data in vivo, it was recently proposed that during the course of PHP1A, PTH excess together with the defective Gs-mediated hypertrophy inhibition result in promotion of the Gq-mediated chondrocytes hypertrophy favoring short stature along with accelerated bone age.[7] Our patient showed a relatively short stature, whereas his bone age was not accelerated but rather matched the chronological age.
Besides the fact that PHP1A is hallmarked by hypocalcaemia, it is now known that serum calcium-phosphorus abnormalities due to PTH resistance are absent at birth and develop over time.[2] A period of up to 4.5 years is required after the increase in serum PTH levels in order to detect the onset of serum hyperphosphataemia and subsequent hypocalcaemia.[8] Our patient, with his novel molecular profile, represents one of these cases of slow progression, where mild PTH resistance is accompanied by normocalcaemia until the age of our follow-up, similar to several other cases.[9-12]
The genetic cause of PHP1A lies on a locus with imprinting phenomenon, which is explained by the tissue-specific monoallelic expression of different parental alleles of Gsα.[13] To date, there are more than 340 different GNAS mutations, which are causative of PHP1A.[14] These defects are reported as maternally inherited in 50% of the cases, whereas the other 50% are de novo allelic changes.[15] According to current guidelines, for each identified de novo pathogenic variant, the parental germline mosaicism should be considered.[2]
In conclusion, we identified a novel heterozygous mutation in the GNAS gene leading to PHP type 1A (iPPSD2) with normocalcaemia that expands the spectrum of known GNAS mutations related to the disease. This case report highlights the importance of high clinical suspicion in cases with incomplete PHP phenotype. Diagnosis of PHP in affected individuals will promote preventive guidance, treatment of possible endocrine defects, and further research into the underlying natural history and aetiology of the disease.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print