INTRODUCTION
As mineralized tissues, bone and dentin have some similar characteristics. The cells of both tissues secrete extracellular matrix (ECM). Odontoblasts and osteoblasts deposit a non-mineralized, type I collagen-rich matrix—called predentin and osteoid, respectively.[1] In bone, inorganic ECM, comprising 65% of the matrix, mainly consists of hydroxyapatite that forms as a result of interactions between minerals and the matrix. Dentin ECM is more mineralized than bone at 72%. The organic ECM, comprising 35% of the matrix, contains collagenous proteins (90%) and non-collagenous proteins (10%).[2] The collagenous portion is mainly constituted of type I collagen, with trace amounts of type III and V collagen. The non-collagenous proteins can be classified into γ-carboxyglutamate-containing proteins, proteoglycans, glycoproteins, and small integrin-binding ligands N-linked glycoproteins (SIBLINGs). The SIBLING family of proteins includes bone sialoprotein (BSP), dentin matrix acidic phosphoprotein 1 (DMP1), dentin sialophosphoprotein (DSPP), osteopontin (OPN), and matrix extracellular phosphoglycoprotein (MEPE). SIBLING proteins are believed to play key biological roles; share an arginine-glycine-aspartic acid (RGD) sequence that facilitates cell attachment, migration, and differentiation; and trigger intracellular transduction by binding to cell surface receptors such as integrin.[1]
BSP is expressed in mineralized tissues, promotes bone formation, and initiates matrix mineralization. It acts as a nucleator of the initial apatite crystal and later as an inhibitor directing the growth of the crystal.[3] BSP is expressed considerably less in dentin than bone, as it constitutes about 1% of the total non-collagenous proteins in dentin. While not much is known about BSP’s role in dentin formation, it is suspected to intensify collagen fibrillation and promote crystal nucleation, with its RGD sequence favoring cell adhesion to the ECM.[4] DSPP is cleaved rapidly into dentin sialoprotein (DSP) in the N-terminus, dentin glycoprotein in the middle, and dentin phosphoprotein (DPP) in the C-terminus. The DPP domain is the DSPP portion that contains the RGD sequence and activates mitogen-activated protein kinase (MAPK) and SMAD by interacting with integrin. The DSP domain acts as a ligand in interaction with integrin B6 and participates in intracellular signaling to promote dental mesenchymal cell differentiation.[5] DSP is believed to be responsible for the initiation of dentin mineralization, while DPP is involved in the maturation of mineralized dentin. Just as a small amount of BSP is found in dentin, DSP is found in rat long bones about 1/400 of dentin.[1] Therefore, despite their names, it would be inappropriate to think of BSP or DSP as bone- or dentin-specific, respectively. DMP1—expressed in bone, dentin, and cementum—regulates phosphate metabolism and promotes bone mineralization.[6] It is believed that the role of DMP1 overlaps with that of DSPP. DSPP is known to participate in the matrix mineralization process of dentinogenesis and osteogenesis. However, in addition to mineralization, DMP1 participates in gene regulation, fibroblast growth factor (FGF) 23 dynamics, and phosphorus metabolism.[6] Additionally, DMP1 was found to go through proteolytic processing, and its fragments play separate roles, as the N-terminus is found in unmineralized predentin, while the C-terminus is found in mineralized dentin.[7] OPN is expressed in osteoblasts, odontoblast, and osteocytes. It regulates bone formation, mineralization, and turnover by inhibiting apatite formation and growth through osteoclast and osteoclastogenesis activity.[1,8] OPN also is believed to anchor osteoclast to the bone surface during bone resorption.[9] MEPE, expressed in osteocytes and dentin, binds strongly to hydroxyapatite minerals in the ECM. MEPE’s involvement in mineralization inhibition and phosphate metabolism regulation was confirmed, with MEPE-null mice showing increased bone mass, while MEPE-overexpressed mice showing decreased bone mass.[10] SIBLING proteins are found not only in mineralized tissues but also in salivary glands and prostate tumors.[11] More research is needed to identify the mechanisms of action and interactions among members of the SIBLING protein family, especially in tissues in which a particular protein does not seem to play a predominant role.
The fact that bone and dentin are both mineralized tissues makes them vulnerable to hereditary mineralization defects, such as hypophosphatemic rickets and osteogenesis imperfecta. However, bone and dentin have morphological differences that make them react differently to injury. In this review article, we describe some of the similarities and differences in bone and dentin, with an emphasis on their healing processes.
HEREDITARY MINERALIZATION DEFECT
Because of the similarities in dentin and bone, some disorders that disrupt mineralization result in abnormalities in both types of tissue. Two such genetic disorders are hypophosphatemic rickets and osteogenesis imperfecta.
X-linked hypophosphatemic rickets—the most commonly inherited pattern among hypophosphatemic rickets—is from a mutation in the phosphate-regulating gene with homologies to endopeptidases on the X chromosome (PHEX) gene. PHEX is involved in calcium and phosphate metabolism and is expressed by osteoblasts, osteocytes, and odontoblast. PHEX interacts with MEPE and protects the C-terminal site from cleavage, which is immediately upstream of the acidic serine-and aspartate-rich motif (ASARM).[12] When the PHEX gene is mutated, a peptide in the MEPE’s ASARM region is liberated and inhibits osteoblast bone morphogenetic proteins 2 (BMP2)-mediated mineralization.[13] The consequences of this mineralization inhibition are manifested clinically in both bone and dentin through leg bowing, short stature, high bone mass, and spontaneous infectious tooth abscesses.[14]
Osteogenesis imperfecta is an autosomal dominant disorder caused by a mutation of the COL1A1 or COL1A2 genes that encode type I collagen, and, less commonly, CRTAP or LEPRE1 genes.[14] Type I collagen mutations significantly affect bone flexibility and strength, giving this disorder the nickname, brittle bone disease.[15] Osteogenesis imperfecta’s type I, the most common type of the disease, has milder clinical features such as fragile bones, blue sclera, early deafness, dentinogenesis imperfecta type I, growth deficiency, and joint laxity. Dentinogenesis imperfecta is characterized by yellow or gray tooth discoloration, rapid attrition, bulbous crowns, and early pulp obliteration. Additionally, in dentinogenesis imperfecta type I patients, collagen fibril diameter is reduced and dentin hardness is impaired, highlighting the important role collagen plays in dentin stability.[16]
BONE
Bone formation is followed by either intramembranous or endochondral ossification. The bone deposition of the former is completed directly through osteoblasts, which differentiate from mesenchymal progenitors through transcription factor cbfa/runx2; the latter of the bone formation is followed by osteochondroprogenitor. Although the 2 ossification processes stem from different sources, they eventually merge at final bone depositing steps - proliferation, matrix maturation, and mineralization - by preosteoblasts. Some osteoblasts are also differentiated into osteocytes, which are located in narrow interconnecting passage ways within bone matrix - bone canaliculi.[17] Nevertheless, the process by which osteoblasts are fixed into bone matrix and molecular mechanism behind its conversion into osteocytes are yet to be discovered.[18]
Osteoclasts are multinucleated cells that are responsible for bone resorption. They are of hematopoietic origin. Macrophage colony-stimulating factor (M-CSF) and the receptor activator of nuclear factor-κB ligand (RANKL) are responsible for the differentiation and activity of osteoclasts. M-CSF and RANKL bind to their respective receptors, c-Fms and RANK, that are expressed on the cell surface of osteoclast progenitor cells, They stimulate differentiation and regulation of osteoclasts via intracellular signaling pathway system.[19]
1. Structure
Bone is organized in a hierarchical order, from nanometer- to millimeter-sized structures.[20] This well-constructed organization produces the stiffness and toughness that provides mechanical support to the bone. The way in which major components are organized vary and is a differentiating factor between members of the bone family.[21] Bone structure is further complicated by the hierarchical levels of bone organization.
Bone is a compound of 3 major components. Approximately 65% of bone (by weight) is mineral, 25% is organic materials, and 10% is water. Carbonated apatite, the only mineral in bone, is found as extremely small crystals in a highly ordered arrangement.[22,23] Of the 25% of bone that is organic material, 90% is composed of type 1 collagen and the remainder of non-collagenous proteins (NCPs).[20] While the collagen’s basic structure is a triple-helix form, each arrangement may vary, as they tend to join with one another.[20,24] Depending on its location within the bone tissue, this property also can influence mechanical stiffness. Although the organic materials in bone include more than 200 NCPs, there is not sufficient evidence showing they provide mechanical support in the tissue.[25] Lastly, water fills in the gaps between each triple-helix fibril, as well as between fibrils and fibers.[26] The relative proportions of these major components remain nearly the same, except that the mineral components increase overtime at the expense of water.[21]
The layers of crystals positioned within the collagen fibril continue to grow until they fuse together and compress the triple helix model to form lengthy sheets.[27] Once mineralized by crystals, collagen fibrils are grouped in bundles.[28] Although these bundles are positioned along their lengths, their overall structure varies from place to place, because they tend to fuse with other mineralized collagen fibers to form an extended framework.[29] These structures include parallel-, woven fiber-, and plywood-like structures.[21]
Bone is known to undergo internal remodeling of itself, and the cells playing distinct roles that balance each other in this process. Osteoclasts are bone-resorbing cells typically found on the bone surface that demineralize the matrix by creating an acidic environment. Once osteoclasts hollow out a big tunnel, it is refilled with layers of mineralized collagen fibrils and osteoblasts depositing lamellar bone. Lamellae make up the bone’s basic metabolic unit, which is called an osteon. Within an osteon are osteocytes, which are mature osteoblasts entrapped in the matrix they secreted. This property is why the bone cross-section is described as having an onion-like structure, as it contains layers of lamellae surrounding a central blood vessel.[21]
Both compact and trabecular bones consist of layers of lamellae, which are classified into 3 distinct types: concentric, interstitial, and circumferential. Concentric lamellae surround an osteon. Within each osteon is a Haversian canal, where capillaries are located. Haversian canals are connected to each other by Volkmann’s canals, which run horizontally to provide a network throughout the bone. Osteocytes communicate with each other through a network of long, cytoplasmic extensions in canaliculi. Interstitial lamellae are previously concentric lamellae that have been pushed outward as a result of bone remodeling to fill the space between concentric lamellae. Circumferential lamellae from the perimeter of the bone, bordering periosteum on the outer side and bone marrow on the inner side.[30]
2. Repair and regeneration process
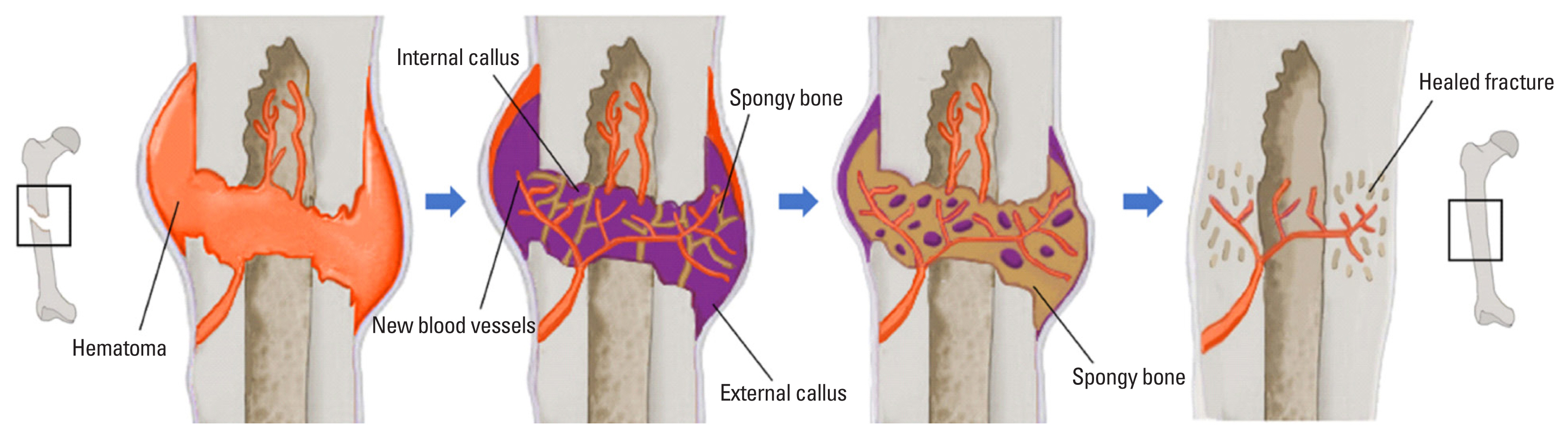
Bone shows unique reparative characteristics following damage, mainly fractures. There are 2 types of bone healing: primary and secondary (Fig. 1). Primary bone healing is only possible when 2 fragmented parts are secured together by orthopedic fixative methods. This type of healing does not involve callus formation; rather, bone remodeling by osteoblasts and osteoclasts results in direct bone healing.[31,32]
The most common type of bone healing is secondary bone healing, which follows a slight movement at the fracture site.[33] Secondary bone healing takes place in four overlapping stages.[34-36] Following the injury, blood vessel and musculoskeletal system disruption occurs concurrently. Therefore, the secondary bone healing process is initiated through the activation of a coagulation cascade, which leads to hematoma formation. The hematoma serves as a transient framework that facilitates stem cell differentiation, in which these cells later convert into fibrous tissue, cartilage, and bone.[37] Angiogenesis is stimulated at the same time, forming new blood vessels from preexisting ones. These new blood vessels supply nutrients and various cell types including fibroblasts, stem cells, and inflammatory cells.[35] Next, the inflammatory response produces swelling, heat, and pain, along with the secretion of various growth factors and cytokines, which contribute significant roles in the bone healing process.[35,36,38,39]
Hematoma formation ensues, forming soft (internal) callus and hard (external) callus. Tissue forms at a fractured site, which later is replaced by fibrocartilage that hardens into soft callus with calcium hydroxyapatite.[40] Soft callus can be mineralized further via endochondral ossification into a hard callus of woven bone.[33,40] At the same time, a hard callus can form independently at the periosteum via intramembranous ossification that facilitates direct bone formation.[40]
Lastly, bone remodeling replaces the fractured callus with the secondary bone. In this final stage of the healing process, both soft and hard callus are reduced in size, eventually reaching a size similar to that of the pre-fracture state.[40] The vasculature system returns to the normal state, as well.[40]
One of the distinguishing features of bone is its rich vascular network. Bones receive blood from proximal, distal epiphyseal, metaphyseal, diaphyseal, and periosteal arteries.[41] This vasculature supplies tissues with blood, nutrients, growth factors, hormones, cytokines, and chemokines, allowing for effective communication with neighboring tissues.[42,43] The vascular system is restored via 2 main signaling pathways. First, platelets serve as the first responders at the site, binding to subendothelium’s exposed collagen and initiating the coagulation cascade. Subsequent hematoma formation then releases several signaling molecules and angiogenic growth factors. Moreover, the subsequent hypoxic environment that follows the injury initiates an angiogenic pathway for bone regeneration together with osteoblasts.[42,44,45] Osteoblasts in turn affect endothelial cell proliferation, differentiation, and secretion of osteogenic growth factors by endothelial cells.[42,44,45] Once angiogenesis takes place, new vasculatures recruit and control migration of other inflammatory cells and repair cells as described previously.[46-51]
During the bone healing process, the following 3 types of blood vessels are enhanced to increase the blood supply to the fracture site: medullary, periosteal, and osseous arteries.[41,52] The newly generated blood supplies to the callus and cortical bone persist until the medullary blood supply is fully generated, allowing the metabolically active, regenerating callus to be supplied with nutrients and signaling molecules, along with osteoblast/osteoclast precursors.[37,52]
Angiogenesis is mediated by several growth factors and signaling molecules: platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), FGF, BMP, and transforming growth factor-β (TGF-β).[40] It has been reported that VEGF plays a crucial role in the bone-repair cascade by acting as a central mediator for angiogenesis and regulating the activity of these pro-angiogenic factors.[53] VEGF expression is induced by the presence of these growth factors, and cytokines are produced in response to hypoxia in osteoclasts.[37,40] In addition to the recruitment and survival of osteoblasts and osteoclasts, VEGF is involved in the mobilization, recruitment, differentiation, and proliferation of endothelial progenitor cells.[47,49,51, 54-56] VEGF is elevated, especially at the site near the fracture, to exert a potent angiogenic activity at the injury site. When VEGF is inhibited, the angiogenic activity of other pro-angiogenic growth factors is prevented, as well.[54,57]
Damage to the blood vessel underneath and subsequent hematoma formation leads to various systemic and local responses. Systemically, parathyroid hormone, growth hormone, steroids, calcitonin, and vitamin D affect bone metabolism and vascularity.[35] Locally, platelets and the hypoxic environment mediate the healing process.[42,44,45] Various types of inflammatory cells near the injured site produce FGF, TGF, BMP, insulin-like growth factor, PDGF, and VEGF.[34-36,38,58,59]
TGF-β—released by degranulating platelets—promotes bone repair by recruiting and proliferating mesenchymal cells.[60,61] Once the mesenchymal cells differentiate into chondrocytes, osteoblasts, and osteoclasts, their activities (e.g., differentiation, ECM production, and osteoblast/osteoclast coupling) also respond to TGF-β, with these cells also producing TGF-β.[40] Mesenchymal cells at the periphery of the injury differentiate into osteoblasts that lay down osteoids. Osteoids go through further mineralization steps to eventually restore the bone’s original shape, structure, and mechanical properties.[62] Because of these roles, TGF-β is suspected to modulate the activity of FGF-2, which induces angiogenesis and mitogenesis of mesenchymal cells and osteoblasts.[63-65]
DENTIN
For ectomesenchymal cells in the most peripheral layer of dental papilla to differentiate into odontoblasts, it requires cell-ECM-mediated epithelio-mesenchymal interaction. Crown dentin requires interaction with pre ameloblast while root dentin requires interaction with inner enamel epithelium of Hertwig’s epithelial root sheath.[66] In addition, molecular signals from basal lamina such as wnt-10a, TGF-β1, TGF-β2, TGF-β3, FGF-1 are essential for the differentiation of odontoblasts.[67-69]
1. Structure
Primary and secondary dentin is composed of tubular structures. With odontoblast cell bodies located at the periphery of the pulp, their cellular processes cross the predentin and run in canaliculi that traverse the dentin layer up to the dentinoenamel junction, resulting in a structure referred to as dentinal tubules. The tubular nature of dentine gives an unusual degree of permeability to this hard tissue, which can promote carious processes and enhance the pulp’s response to dental restorative procedures.[30] The 2 main layers of dentin are the outermost mantle layer and the circumpulpal dentin (the largest part of the dentin layer). Circumpulpal dentin can be categorized into 2 types: intertubular and peritubular dentin. Intertubular dentin refers to the dentin between tubules, consisting of a tightly interwoven network of type I collagen fibrils in and around which apatite crystals are deposited. Fibrils are arranged randomly in a plane at roughly right angles to the dentinal tubules. Its ground substance consists of NCPs and some plasma proteins. Peritubular dentin delimits the dentinal tubule. This dentin is poor in collagen and more mineralized than the rest of the dentin.[30] The arrangement of collagen fibrils in dentin, specifically in the intertubular dentin, can be described as radial fibril arrays. With the collagen fibrils in the plane parallel to the surface at which dentin formation takes place in the pulp cavity, the dentin structure is highly anisotropic (i.e., its physical properties vary depending on directionality). However, within the collagen fiber, the fibril bundles are randomly and poorly oriented. Furthermore, the crystal layers within the fibril bundles also are randomly placed. This random orientation of crystals within dentin collagen fibers provides isotropic elastic and hardness properties. Despite the isotropic elasticity of dentin structures, the fracture properties of dentin are anisotropic, in which the crack follows the plane of mineralized dentin collagen fibrils.[21]
2. Repair and regeneration process
The major clinical consequence of the difference between bone and dentin is how they heal in response to injury. While the bone healing process is regenerative, the dentin’s healing process is an imperfect repair mechanism that results in a structure that differs significantly from the original tissue.[70]
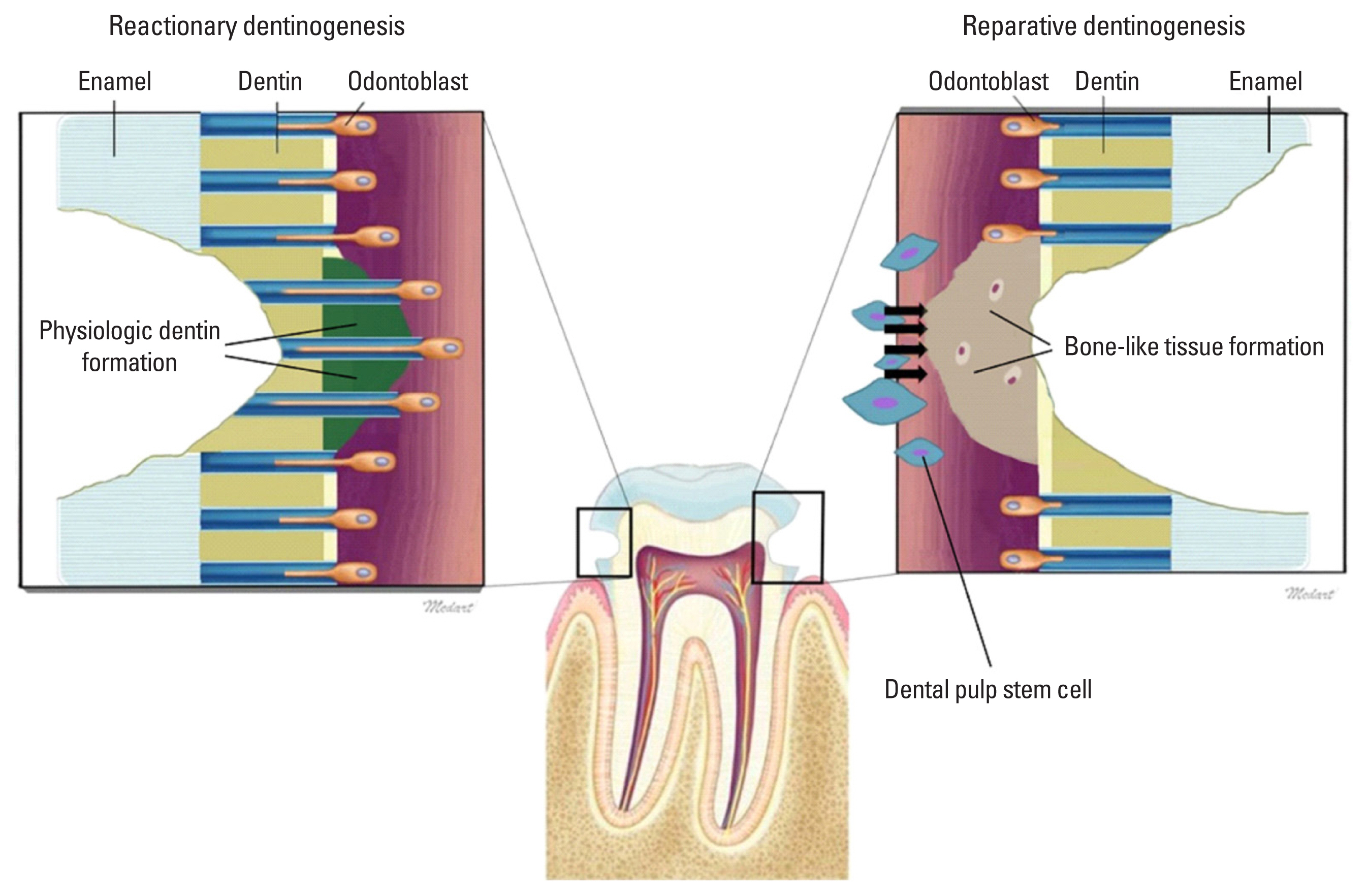
Tertiary dentin—formed in response to irritating stimuli—can be classified into reactionary and reparative dentin (Fig. 2). Reactionary dentin is formed after a milder stimulus in which primary odontoblasts survive the injury. In such cases, growth factors from the matrix reach the existing odontoblasts at the injury site to increase the secretion rate of the matrix. Down-regulation of odontoblast secretion by autophagy during secondary dentinogenesis is up-regulated again during reactionary dentinogenesis via the p38-MAPK pathway. This happens probably through a combination of bacterial stimulants and the release of fossilized growth factors and bioactive molecules derived from demineralized primary and secondary dentin matrix. Upon histological observations, it was found that reactionary variants—classified by the continuity of the tertiary dentin with the secondary dentine—had sparsely distributed tubules that ran irregularly.[70]
In contrast, reparative dentin is formed in response to a stronger stimulus that kills the primary odontoblasts. This type of dentin requires molecular signals from dentin matrix-derived growth factors to induce migration, proliferation, and differentiation of the progenitor cells to odontoblast-like cells, which then replace the dead odontoblasts and secrete a new dentin matrix. Three possible theories explain the origin of these odontoblast-like cells. First, it has been suggested that one of the daughter cells is positioned at the dental basal membrane during the pre-odontoblasts’ last cell division, right before the terminal differentiation. When the inductive signals are given (e.g., bacterial stimulants or fossilized growth factors released from the demineralized matrix diffuse into the area where these daughter cells are located), these cells differentiate into the odontoblasts. In order to become odontoblast-like cells during the tertiary dentinogenesis, the daughter cells migrate into the Höhl layer of the dental pulp. Second, it has been suggested that the cells in the pulp tissue (pericytes, fibroblasts, and endothelial cells, which can be found in the pulp’s bipolar zone), may convert into undifferentiated mesenchymal stem cells. These cells then re-differentiate into odontoblast-like cells. A definitive mechanism explaining these steps has not been found. Third, the existence of multipotent adult stem cells has been suggested. While these cells may play a key role in tertiary dentinogenesis, it is not certain whether they migrate to the injury site via the pulp tissue vasculature or have a developmental derivation from dental papilla.[71]
Reparative dentin can be classified into either osteodentin or orthodentin. The orthodentin type has tubules but no cell inclusions.[4] Poorer repairs lead to the osteodentin type, which—unlike a typical odontoblast cell—resembles bone osteocytes that are entrapped in the reparative dentin with a strong expression of BSP.[72] It was found that when there is a cavity large enough to result in the absence of odontoblasts near the injury site, an amorphous, atubular calcified tissue (not continuous with the primary or secondary dentin structure) was laid down. It is hypothesized that this calcified tissue, uncharacteristic of dentin, may result from the proliferation of fibroblasts, the predominant cell type in the ECM of the dental pulp, may outcompete that of the progenitor cells following the death of primary odontoblasts.[70] Generally, when human dentin pulp stem cells are induced to differentiate into odontoblast-like cells, they are involved in reparative dentin formation. When carious insults demineralize the dentin, growth factors embedded in the dentin matrix diffuse into the pulp stem cell layers. One of the biomolecules involved in signaling the pulp stem cells is TGF-β. TGF-β is known to be heavily involved in the wound-healing process, where it contributes to fibrosis formation. We posit that TGF-β is the key molecule signaling the pulp stem cells to differentiate into the fibroblasts that creates reparative dentin. Interestingly, it has been suggested that CPNE7 molecules induce differentiation of odontoblast-like cells from mesenchymal cells of dental or non-dental origin.[73] Current research on CPNE7 molecules has shown that when pulp stem cells are induced by CPNE7, they not only have the ability to differentiate into the odontoblast-like cells, but they also form the dentin structure that is similar to reactionary dentin.[74] From this result, we can infer that the CPNE7 molecule is essential in making the physiologic reactionary dentin with dentinal tubular structures during the process of tertiary dentinogenesis. However, the exact biological mechanism of this process is unknown.
CONCLUSIONS
In this article, we described similarities and differences in bone and dentin, emphasizing the healing process of each. Bone and dentin share similar characteristics in their composition, biomarkers, and clinical manifestations in the case of mineralization defects. There has been a shift in thought that some of the matrix-derived proteins traditionally described as either dentin- or bone-specific should not be described as such since it was recently determined that such proteins are expressed in both tissues, albeit at a lower level. Some of the key differences are the presence of dentinal tubules, vascularity, and the ability to remodel throughout the lifespan. Due to these morphological differences, we determined that bone has a more effective regenerative process than dentin.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print