Myostatin Inhibitors: Panacea or Predicament for Musculoskeletal Disorders?
Article information

Abstract
Myostatin, also known as growth differentiation factor 8 (GDF8), is a transforming growth factor-β (TGF-β) family member that functions to limit skeletal muscle growth. Accordingly, loss-of-function mutations in myostatin result in a dramatic increase in muscle mass in humans and various animals, while its overexpression leads to severe muscle atrophy. Myostatin also exerts a significant effect on bone metabolism, as demonstrated by enhanced bone mineral density and bone regeneration in myostatin null mice. The identification of myostatin as a negative regulator of muscle and bone mass has sparked an enormous interest in developing myostatin inhibitors as therapeutic agents for treating a variety of clinical conditions associated with musculoskeletal disorders. As a result, various myostatin-targeting strategies involving antibodies, myostatin propeptides, soluble receptors, and endogenous antagonists have been generated, and many of them have progressed to clinical trials. Importantly, most myostatin inhibitors also repress the activities of other closely related TGF-β family members including GDF11, activins, and bone morphogenetic proteins (BMPs), increasing the potential for unwanted side effects, such as vascular side effects through inhibition of BMP 9/10 and bone weakness induced by follistatin through antagonizing several TGF-β family members. Therefore, a careful distinction between targets that may enhance the efficacy of an agent and those that may cause adverse effects is required with the improvement of the target specificity. In this review, we discuss the current understanding of the endogenous function of myostatin, and provide an overview of clinical trial outcomes from different myostatin inhibitors.
INTRODUCTION
Myostatin, also known as growth differentiation factor 8 (GDF8), is a transforming growth factor-β (TGF-β) family member that potently inhibits skeletal muscle development [1]. The biological function of myostatin became evident when mice homozygous for a deletion of myostatin gene exhibited a dramatic increase in skeletal muscle mass, with individual muscle groups enlarging to approximately twice their normal size [1]. Remarkably, the amino acid sequence of myostatin and its function are extremely well conserved across different species, and a hypermuscular phenotype resulting from naturally-occurring mutations in the myostatin gene was detected in humans [2], cattle [3], sheep [4], and dogs [5]. In addition to its effect on skeletal muscle, myostatin has been demonstrated to play a significant role in the regulation of bone metabolism by suppressing bone formation [6] and stimulating bone resorption [7]. Indeed, myostatin null mice represent not only a doubling of muscle mass, but also enhanced bone mineral density (BMD) and bone regeneration [6,8].
The establishment of myostatin as a robust negative regulator of muscle and bone mass has designated myostatin as an attractive therapeutic target for various musculoskeletal disorders. In fact, numerous myostatin-inhibiting pharmacological agents have been developed, and many of them have progressed to human trials and are currently under evaluation (Table 1). What must be considered is the fact that most myostatin inhibitors also block the activity of other closely-related members of the TGF-β family [9-12], raising the possibility of undesired side effects. In this regard, our group has recently demonstrated that overexpression of follistatin (FST), an endogenous antagonist of myostatin, increases skeletal muscle mass by suppressing the activity of myostatin, but diminishes BMD and induces bone fractures likely through binding and repressing the activity of GDF11, a myostatin paralog that promotes osteogenesis in contrast to myostatin, emphasizing that the opposing roles of myostatin and GDF11 must be carefully considered when generating myostatin inhibitors for therapeutic intervention [6]. In this review, we will first describe the current understanding of the mechanism of myostatin function, and subsequently present an overview of the clinical progress, efficacy, and side effects of different myostatin inhibitors. Finally, we will discuss future perspectives and challenges regarding the development of effective myostatin-targeting therapeutics with minimal adverse effects.

Clinical trials of various myostatin-targeting drugs
THE PHYSIOLOGY OF MYOSTATIN
Since the discovery of myostatin in 1997, insights on the molecular mechanism of its action on growth and maintenance of skeletal muscle and other tissues have substantially improved, catalyzing the development of novel myostatin-targeting strategies (Fig. 1). In the following section, endogenous functions, activation processes, and signaling pathways of myostatin will be discussed.
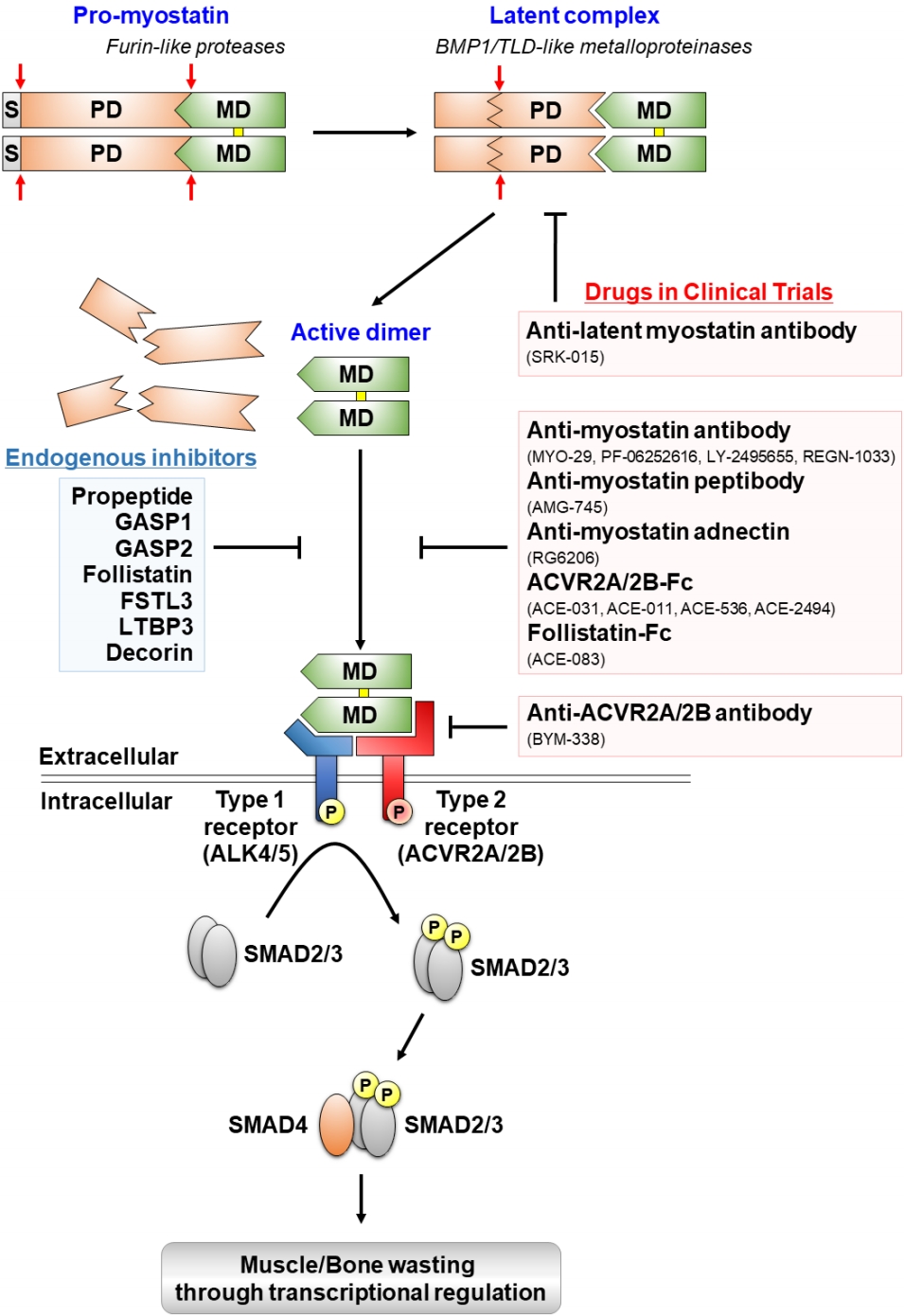
Proteolytic processing, extracellular regulation, and signaling mechanisms of myostatin and myostatin-targeting drugs in clinical trials. Myostatin is first synthesized as a precursor molecule (pro-myostatin) that undergoes proteolytic processing to produce the biologically active molecule. After removal of the signal peptide (S), pro-myostatin is cleaved by a furin-like protease to form a latent complex, leaving the mature domain (MD) non-covalently attached to the prodomain (PD). The latent complex is further cleaved by a bone morphogenetic protein 1 (BMP1)/ tolloid (TLD)-like metalloproteinase to generate the mature, disulfide-linked (marked in yellow), dimer that initiates signal transduction. The mature myostatin ligand binds to constitutively phosphorylated activin type 2A and 2B receptors (ACVR2A/2B) that subsequently recruit and phosphorylate (P) activin receptor-like kinase 4 and 5 (ALK4/5), leading to phosphorylation and activation of Smad2/3. Finally, activated Smad2/3 assembles with Smad4 and translocates to the nucleus to regulate expressions of target genes associated with muscle and bone homeostasis. Endogenous inhibitors of myostatin and myostatin-targeting drugs in clinical trials are indicated by blue and red boxes, respectively.
1. Endogenous functions of myostatin
The primary biological function of myostatin became apparent when the disruption of myostatin gene in mice resulted in a striking increase in skeletal muscle mass throughout the body. More specifically, analysis of myostatin null mice revealed that these mice exhibit both an increase in muscle fiber number (hyperplasia) and muscle fiber size (hypertrophy) [1]. Because muscle growth during postnatal periods is mainly caused by muscle hypertrophy [13], muscle hyperplasia observed in myostatin knockout animals suggests that myostatin suppresses the function of muscle progenitor cells and myogenesis during embryonic development. Indeed, myostatin is strongly expressed in developing somites [1], and its overexpression in embryonic chicken somites impeded muscle development through depletion of muscle progenitor cells [14]. Meanwhile, postnatal inactivation of myostatin through administration of monoclonal anti-myostatin antibody in adult mice also exhibited muscle hypertrophy with enhanced force production, highlighting the direct role of myostatin in regulating adult muscle fiber size [15]. Similarly, systemic overexpression of myostatin in adult mice resulted in a substantial muscle loss similar to that observed in human cachexia [16]. Moreover, a notable elevation in myostatin levels, which significantly correlated with a reduction in muscle fiber size and lean mass, were detected both in patients with disuse muscle atrophy associated with osteoarthritis of the hip [17] and subjects who underwent prolonged bed rest [18].
In addition to its role in controlling muscle mass, myostatin has also been actively implicated in regulating bone metabolism and regeneration. For instance, multiple studies have shown that mice lacking myostatin exhibit elevated BMD in the whole body [6,19], limb [20], spine [6], and jaw [21]. These mice also display significantly increased mechanical strength of fracture callus and bone volume after osteotomy of the fibula, which are associated with upregulation of bone morphogenetic protein 2 (BMP2) [8]. Also, transgenic mice overexpressing myostatin propeptide, which binds and inhibits the active myostatin ligand, exhibit elevated BMD [22]. In a transgenic mouse model of rheumatoid arthritis (RA), in which human tumor necrosis factor-α is overexpressed, deficiency of myostatin or its inhibition through antibody led to a significant amelioration of arthritis severity, indicating myostatin as a potent therapeutic target to prevent joint destruction in RA [7]. Furthermore, genetic studies in humans demonstrated that myostatin gene polymorphisms are associated with peak BMD [23], further supporting the involvement of myostatin in regulating bone homeostasis. Notably, the impact of myostatin on bone may be both direct and indirect through changes in muscle mass. While an indirect effect of myostatin on bone was evidenced by improvements in bone strength due to increased muscle mass in myostatin null mice [24], direct effects were confirmed by studies showing that myostatin inhibits osteoblast differentiation [6,25,26] and stimulates osteoclast formation [7], thus exerting negative effects on bone mass through reduced bone formation and increased resorption.
Finally, myostatin has been suggested to exert profound effects on adipogenesis and metabolic function. Indeed, myostatin deficient mice exhibit reduced total body fat in addition to increased muscle mass, and loss of myostatin partially prevented fat accumulation and abnormal glucose metabolism in mouse models of obesity and type 2 diabetes [27]. Also, circulating myostatin levels were reported to be higher in obese humans, and certain polymorphisms in myostatin gene were demonstrated to be associated with obesity [28]. Although further studies are required to clearly establish whether the metabolic effects of myostatin are mediated by its direct signaling to adipose tissues or indirect mechanisms such as changes in muscle mass, the apparent benefits of targeting myostatin pathway seen in animal models of metabolic diseases have greatly supported the potential for myostatin blockade as treatment options for obesity and type 2 diabetes.
2. Activation process of myostatin protein
Myostatin, like other members of the TGF-β family, is first synthesized as a precursor protein and is further cleaved through proteolytic processing events to generate the biologically active signaling domain (Fig. 1) [1]. In detail, after removal of the signal peptide for secretion, a cleavage event occurs at an RSRR site by a furin-like protease, yielding an N-terminal prodomain (propeptide) and a C-terminal mature domain. At this point, the N-terminal and C-terminal domains still remain noncovalently associated to each other to form an inactive latent complex [12], the major circulating form of myostatin in the blood [16]. A full activation of the mature C-terminal dimer requires an additional cleavage at aspartate residue 76 of the N-terminal prodomain by a BMP1/tolloid (TLD)-like metalloproteinase [29]. The active C-terminal dimer can be completely inhibited by adding purified propeptides [12,30].
Targeting the myostatin processing pathway has been actively suggested to be an effective myostatin-inhibiting strategy to promote muscle growth. For example, transgenic mice carrying a mutation in the myostatin RSRR processing site, in which the myostatin precursor protein is resistant to cleavage by furin-like proteases, display enhanced muscular development [31]. Furthermore, mice homozygous for D76A mutation, in which the aspartate residue of the myostatin propeptide is replaced with alanine to prevent the cleavage by BMP1/TLD-like proteinases, exhibit a remarkable increase in muscle mass [32]. Likewise, injection of D76A mutant myostatin propeptide, through binding and inhibiting the mature myostatin molecule, significantly enhanced muscle growth in mice [29], and overexpression of myostatin propeptide using recombinant viral vector also effectively induced muscle hypertrophy in aged mice [33], highlighting the potential for myostatin propeptide as a promising therapeutic option for the treatment of muscle loss-related diseases.
3. Molecular mechanism of active myostatin signaling
The active myostatin dimer initially binds to the activin type 2 receptors (ACVR2A or ACVR2B), featuring a higher affinity for ACVR2B than ACVR2A, and subsequently forms a heteromeric receptor complex with type 1 receptors, activin receptor-like kinase 4 (ALK4) or ALK5, to stimulate the intracellular signal transduction via phosphorylation of Smad2 and/or Smad3 [34]. Myostatin-induced phosphorylation of Smad3 interferes with the activity of MyoD, a master transcription factor in myogenesis, impairing myoblast differentiation [35]. Furthermore, Smad2/3 phosphorylation by myostatin triggers atrophy in adult muscle fibers partially through upregulation of an ubiquitin ligase atrogin1 [36]. In addition, through a Smad2-dependent elevation of nuclear factor of activated T cells 1, myostatin was recently described to directly accelerate the formation of osteoclasts from bone marrow-derived macrophages, increasing bone resorption [7].
One of the most potent ways to inhibit myostatin signaling was demonstrated using the soluble form of ACVR2B receptor. For instance, injection of ACVR2B-Fc fusion protein to wild-type mice induced a dramatic increase (40%-60%) in muscle mass in just 2 weeks [37], an effect greater than that obtained after treatment with D76A myostatin propeptide or monoclonal anti-myostatin antibody. Similarly, administration of ACVR2B-Fc fusion protein to a mouse model of osteogenesis imperfecta (OI) caused significant increases in both bone and muscle mass that were close to those seen in wild-type mice [38]. Importantly, the soluble ACVR2B receptor exhibited anabolic effects even in myostatin null mice, indicating the existence of other TGF-β family members that function to limit muscle growth [37].
The activity of mature myostatin molecule is regulated by multiple extracellular binding proteins including FST, FST-like 3 (FSTL3/FLRG), growth and differentiation factor-associated serum protein 1 (GASP1), GASP2, latent TGF-β binding protein 3 (LTBP3), and decorin [39]. The importance of these inhibitory proteins in controlling myostatin activity is well-demonstrated through substantial muscle hypertrophy and enhanced muscle regeneration observed in mice overexpressing these proteins. Specifically, mice overexpressing FST [12], FSTL3 [40], GASP1 [41], GASP2 [42], or LTBP3 [43] exhibit a hypermuscular phenotype, while decorin gene transfer in mice leads to an improved muscle regeneration [44], consistent with the characteristics present in myostatin null mice. In contrast, mice lacking FST [45], GASP1, or GASP2 [46] display reduced muscle mass and impaired muscle regeneration, effects corresponding to hyperactivity of myostatin. Among multiple myostatin regulatory proteins, FST promoted the greatest improvement muscle size and function when delivered to normal and dystrophic mice [47], leading FST-based therapies to clinical trials (Table 1). Notably, FST antagonizes not just myostatin, but also other TGF-β family members such as activins, GDF11, and BMPs, and the dramatic anabolic effects of FST was reported to result from its inhibition of multiple ligands with similar biological activity to myostatin [40]. There are 3 FST isoforms, including FST288, FST303, and FST315, all of which contain an N-terminal domain and 3 FST domains (FSD1-3) [48,49]. Unlike the other 2 isoforms, FST288 lacks a highly acidic C-terminal tail, exhibits a high affinity for heparin-sulfated proteoglycans, and is tissue-bound, allowing FST288 to act locally. On the other hand, FST315, which accounts for the majority of FST mRNAs, is the predominant isoform found in serum, while FST303 is gonad-specific [48,49]. Among the 3 isoforms, FST288 presents the highest affinity for activins [48].
MYOSTATIN INHIBITORS: TYPES, THERAPEUTIC USES, AND SIDE EFFECTS
Based on the well-established role of myostatin in controlling muscle and bone mass, an intense effort has been directed at targeting this pathway to provide therapeutic benefits under various physiological and pathological conditions including, but not limited to, muscle degenerative diseases, cachexia, sarcopenia, osteoporosis, and type 2 diabetes. Multiple myostatin-targeting strategies such as neutralizing antibodies, myostatin propeptides, soluble ACVR2A or ACVR2B receptors, and naturally-occurring antagonists (FST) have been developed, and those that have progressed into clinical trials are summarized in Table 1. It is important to note that most myostatin inhibitors also interact with other closely-related members of the TGF-β family (GDF11, activins, and BMPs), and while inhibition of these ligands has been reported to induce greater muscle hypertrophy than targeting myostatin alone [40], it can also result in serious adverse effects in other tissues [6,50]. In the following section, the clinical progress of different myostatin inhibitors, their candidate targets, and potential side effects will be discussed.
1. A variety of myostatin inhibitors in different clinical settings
1) Bone-loss disorders
Targeting myostatin emerged as an attractive strategy to prevent bone loss and fractures in addition to promoting muscle growth. Indeed, evidence from both animal and human studies suggests that myostatin exerts a critical impact on bone metabolism and its inhibition may be beneficial for bone turnover [8,22,23]. The fact that tight coupling exists between muscle and bone and that decreased bone development is observed in various myopathies such as Pompe disease [51], spinal muscular atrophy (SMA) [52], and Duchenne muscular dystrophy (DMD) [53], has further underscored the therapeutic potential for myostatin inhibitors as a novel agent to improve musculoskeletal function. The potent anabolic effect of myostatin inhibition on musculoskeletal tissues is supported by data showing that ACVR2B-Fc injection enhances both bone and muscle mass in wild-type and OI mouse models [38]. Likewise, systemic delivery of ACVR2A-Fc increased bone formation, bone mass, and bone strength in both normal and ovariectomized mice [54].
Sotatercept (ACE-011), developed by Acceleron Pharma to treat osteoporosis, is a fusion protein in which the extracellular domain of ACVR2A is linked to a human IgG Fc domain. Notably, biweekly subcutaneous injection of ACE-011 (10 mg/kg) for 3 months led to significant increases in bone volume (93%) and bone formation rate (166%) in cynomolgus monkeys [55]. Similarly, a single injection of ACE-011 (0.01-3 mg/kg) resulted in a sustained dose-dependent elevation in serum levels of bone-specific alkaline phosphatase (BsALP), a biomarker of bone formation, and a dose-dependent reduction in levels of C-terminal telopeptide of type I collagen and tartrate-resistant acid phosphatase 5b, which are biomarkers of bone resorption [56]. A substantial increase in BsALP levels and improvement in BMD were also detected in multiple myeloma patients who received 4 monthly doses of ACE-011 [57], indicating ACE-011 as a potentially effective therapy in bone-loss disorders.
ACE-2494, also developed by Acceleron Pharma as a potential therapeutic agent for bone fragility and muscle-wasting diseases, is a soluble ACVR2B receptor modified to minimize vascular side effects induced by BMP9/10 inhibition [50]. In vivo, subcutaneous injections of ACE-2494 significantly enhanced bone and muscle mass in normal mice, and improved bone length and geometry in a mouse model of OI [58]. However, after the phase 1 study in healthy postmenopausal women, ACE-2494 development was discontinued due to an inconsistent profile of anti-drug antibodies.
2) Muscle-wasting diseases
Stamulumab (Myo-29), developed by Wyeth Pharmaceuticals (now Pfizer), is the first myostatin inhibitor to progress to clinical trials. Myo-29 is a monoclonal anti-myostatin anti-body designed to treat patients with Becker muscular dystrophy, facioscapulohumeral muscular dystrophy (FSHD), or limb-girdle muscular dystrophy (LGMD). Despite encouraging results of the preclinical studies [59] and safety profile, Myo-29 failed to show significant improvements in muscle strength or function likely due to high rate of clearance [60], which halted its further development.
Domagrozumab (PF-06252616) is a more recent humanized monoclonal anti-myostatin antibody developed by Pfizer for the treatment of subjects with DMD and LGMD. While Domagrozumab effectively increased muscle volume in cynomolgus monkeys (24% increase from baseline) and muscle weight in mdx mice (23%-26% increase from baseline) [61], and exhibited a good safety profile and slow clearance rate in healthy subjects [62], it failed to demonstrate time improvements in the 4-stair climb test compared to placebo in the phase 2 study involving boys with DMD, leading to the study termination [63].
Landogrozumab (LY-2495655) is another humanized monoclonal antibody targeting myostatin developed by Eli Lilly & Company to treat patients with sarcopenia (phase 2 completed in 2013), elective total hip replacement (phase 2 completed in 2014), and pancreatic cancer cachexia (phase 2 completed in 2016). In patients aged 75 years or older who had fallen in the past year, landogrozumab injection for 20 weeks increased appendicular lean mass by 0.43 kg and improved stair climbing time, chair rise with arms, and fast gait speed [64]. However, landogrozumab treatment in men and women aged over 50 who received a hip replacement failed to reach the primary outcome of an increase in appendicular lean mass after 12 weeks [65]. In patients with pancreatic cancer, landogrozumab also failed to meet the primary endpoint of an improvement in overall survival [66].
Trevogrumab (REGN-1033), developed by Regeneron Pharmaceuticals Inc., is a monoclonal anti-myostatin antibody designed to treat patients with sarcopenia (phase 2 completed in 2015). REGN-1033, which selectively inhibits pro-, latent, and mature myostatin without binding GDF11 or activin A, effectively enhanced skeletal muscle mass and improved isometric force production in young and aged mice [67]. Efficacy evaluations of the completed phase 2 clinical trial with the primary endpoint of percent change in lean body mass in patients with sarcopenia are currently ongoing.
SRK-015, recently designed by Scholar Rock for the treatment of SMA, is a monoclonal antibody that inhibits the activation of myostatin. SRK-015 binds specifically to pro- and latent form of myostatin, but not mature myostatin, and therefore, unlike most myostatin-targeting biologics, does not cross-react with other members of the TGF-β family (GDF11, activins, and BMPs) that share high degree of similarity in their mature domains [68]. In vivo, SRK-015 analogs effectively improved muscle mass and function in mouse models of SMA [69] and prevented muscle atrophy induced by dexamethasone treatment [68]. SRK-015 is currently being investigated in a phase 2 clinical trial in patients with SMA.
PINTA-745 (AMG-745) is a novel anti-myostatin peptibody (a myostatin-neutralizing peptide fused to a human IgG Fc domain) originally developed by Amgen. While subcutaneous injection of PINTA-745 significantly increased skeletal muscle mass in mouse models of chronic kidney disease [70] and stroke [71], the phase 1/2 clinical trial in patients with end-stage renal disease with protein energy wasting failed to meet the primary endpoints, or an improvement in lean body mass from baseline measured by dual energy X-ray absorptiometry, leading to discontinuation of the drug’s further development.
RG6206 (RO7239361, BMS-986089) is an anti-myostatin adnectin (a genetically engineered variant of the 10th type 3 domain of human fibronectin) coupled to a human IgG Fc domain originally designed by Bristol-Meyers Squibb (later licensed to Roche) to treat patients with DMD [72]. A phase 2/3 clinical trial evaluating the safety, efficacy, and tolerability of BMS-986089 in ambulatory boys with DMD (estimated to be completed in late 2020) was recently discontinued by Roche after the analysis of preliminary results showing that the study was unlikely to reach its primary endpoint of changes in the North Star Ambulatory Assessment score.
In addition to antagonizing the myostatin ligand, targeting its receptors to block the signaling transduction pathway has also been widely explored as a treatment strategy for muscle-loss disorders. For instance, bimagrumab (BYM-338), developed by Novartis in collaboration with MorphoSys AG, is a monoclonal antibody that binds competitively to activin type 2 receptors (has greater affinity for ACVR2B than ACVR2A [73]), thus preventing the binding of natural ligands (GDF11, activins, and BMPs) to the receptors that initiate downstream phosphorylation of Smad2/3, which is significantly increased in muscle tissues of patients with sporadic inclusion body myositis (sIBM) [74]. In 2013, the Food and Drug Administration (FDA) granted the breakthrough therapy designation to bimagrumab for sIBM. However, while the phase 2 clinical trial in 14 sIBM patients demonstrated increases in thigh muscle volume and 6-min walking distance after a single injection (30 mg/kg) [74], subsequent phase 2/3 clinical trial completed in 2016 failed to show improvement in 6-mintue walking distance, muscle strength, or grip and pinch strength [75], resulting in discontinuation of further development of bimagrumab for sIBM. In a phase 2 clinical trial conducted in subjects older than 65 years with sarcopenia, treatment with bimagrumab (30 mg/kg) for 16 weeks significantly improved thigh muscle volume, gait speed, and 6-min walking distance [76]. However, a further dose range finding study (6 monthly doses of 70, 210, or 700 mg) in patients with sarcopenia resulted in 2 deaths in the highest-dosage group, halting the development of bimagrumab as a treatment for sarcopenia. In a phase 2 clinical trial involving healthy young men who were placed in a cast for 2 weeks to induce disuse atrophy, a single injection (30 mg/kg) of bimagrumab significantly accelerated thigh muscle volume recovery [77]. Furthermore, a phase 2 clinical trial in chronic obstructive pulmonary disease (COPD) patients with cachexia demonstrated a significant increase in thigh muscle volume, but not 6-min walk distance, after 2 injections of bimagrumab (30 mg/kg) [78].
Ramatercept (ACE-031) is a soluble form of ACVR2B developed by Acceleron Pharma for the treatment of DMD. Despite encouraging results of both the animal study [79], in which ACE-031 significantly increased both type 1 and type 2 muscle fiber cross-sectional area in young mice, and phase 1 clinical study in healthy postmenopausal women [80], in which ACE-031 injection (3 mg/kg) significantly increased total body lean mass and thigh muscle volume, subsequent phase 2 clinical trial in patients with DMD was terminated due to occurrence of serious non-muscle-related adverse events including nosebleed, gum bleeding, telangiectasia, and/or erythema [50], halting further development of the drug.
ACE-083, a FST-based fusion protein in which FST291 is coupled to a human IgG Fc domain [81], is an alternative version of ACE-031 developed by Acceleron Pharma for the treatment of FSHD and Charcot-Marie-Tooth disease (CMT). Notably, ACE-083 is designed to act locally through intramuscular administration and its treatment effectively induced localized muscle hypertrophy and focal improvement in force generation without systemic effects in wild-type mice and mouse models for CMT and DMD [81]. In a phase 1 study performed on healthy postmenopausal women, ACE-083 injection (50-200 mg) into muscles resulted in significant targeted muscle growth with no change in strength [82]. Unfortunately, phase 2 clinical trials in patients with FSHD and CMT were recently terminated due to failure in achieving functional improvements, shutting down further development of ACE-083.
Intramuscular gene transfer of rAAV1.CMV.huFollistatin 344 is currently being developed by Nationwide Children’s Hospital and Milo Biotechnology to treat patients with Becker muscular dystrophy, sIBM, and DMD. FST344 is a FST isoform that is cleaved to produce the serum circulating FST315 isoform [48], and when delivered by an AAV1 vector to muscles in cynomolgus monkeys, increases in both muscle size and strength were observed [83]. In a phase 1/2 clinical trial of patients with Becker muscular dystrophy, direct bilateral intramuscular quadriceps injections (3×1011 vg/kg/leg or 6×1011 vg/kg/leg) of rAAV1.CMV.huFollistatin 344 significantly improved 6-min walking distance in 4 out of 6 patients [84]. Likewise, in a phase 1/2 clinical trial of patients with sIBM, a single bilateral intramuscular quadriceps injections (6×1011 vg/kg/leg) of rAAV1.CMV.huFollistatin344 significantly increased 6-minute walking distance [85], showing promise for FST gene therapy for these muscle-wasting conditions. A phase 1/2 study conducted on patients with DMD, in which a higher dose (2.4×1012 vg/kg/patient) of rAAV1.CMV.huFollistatin344 was delivered to gluteal muscles, quadriceps, and tibialis anterior, is also completed and under evaluation.
3) Other disorders (anemia and metabolic diseases)
RAP-011 (murine ortholog of sotatercept/ACE-011) treatment in mice, likely through inhibiting GDF11, a ligand reported to impede terminal erythroid maturation [86,87], induced a rapid increase in hematocrit, hemoglobin (Hb), and red blood cell (RBC) parameters accompanied by stimulation of late-stage erythropoiesis [87]. ACE-011 in a phase 1 clinical trial conducted on healthy postmenopausal women also increased hematocrit, Hb, and RBC levels [56]. These results have evoked a considerable interest in ACE-011, originally developed to treat bone-loss disorders, as a potential therapeutic option for patients with anemia and diseases involving ineffective erythropoiesis. In a phase 2 clinical trial of patients with myelodysplastic syndromes (MDS), ACE-011 effectively decreased transfusion burden in 47% of patients with a high transfusion burden, while elevating Hb levels in 58% of patients with a low transfusion burden [88]. Likewise, in a phase 2 clinical trial involving patients with β-thalassemia, 75% of non-transfusion-dependent patients treated with higher doses resulted in a sustained increase in Hb levels [89]. In addition, preclinical studies in animal models of pulmonary arterial hypertension (PAH) showed consistent efficacy of sotatercept [90], and a phase 2 human trial for the treatment of PAH is currently ongoing.
Luspatercept (ACE-536), a sotatercept-analogous erythroid maturation agent developed by Acceleron Pharma, is a fusion protein that comprises the extracellular domain of ACVR2B coupled to a human IgG Fc domain modified to preferentially bind GDF11 and myostatin over activin A [86]. In phase 2 and 3 clinical trials of patients with β-thalassemia [91], ACE-536 effectively improved erythropoiesis, Hb levels, and reduced RBC transfusion requirements. Based on the results of the phase 3 clinical trial conducted on patients with β-thalassemia, which demonstrated a significantly greater percentage of successful transfusion burden reductions in the luspatercept group than the placebo group [92], ACE-536 was recently approved by the FDA for the treatment of anemia associated with β-thalassemia [93]. A phase 3 clinical trial in patients with MDS is currently ongoing.
Bimagrumab, a monoclonal antibody against activin type 2 receptors, was reported to improve lean mass (2.7%) and insulin sensitivity (20%-40%) while reducing fat mass (7.9%) on subjects with insulin resistance [94], suggesting that bimagrumab may provide a novel treatment approach for the metabolic complications of obesity. Currently, development of bimagrumab as treatment options for obesity and type 2 diabetes is ongoing (phase 2 study is completed under evaluation).
2. Multiple targets of myostatin inhibitors
A major advancement in understanding of the myostatin regulatory mechanism has greatly facilitated the development of promising myostatin inhibitors in the form of antibodies, soluble receptors (ACVR2A-Fc and ACVR2B-Fc), and natural binding proteins (FST), which have been introduced to clinical trials for application in a wide range of disease settings. Importantly, due to the high degree of similarity between the receptor-recognition surfaces of mature myostatin and other members of the TGF-β family [95], majority of myostatin-targeting agents cross-react with other TGF-β family members. Specifically, GDF11 and myostatin share 89% sequence identity within their mature signaling domain [39], and as a result, myostatin antibodies cross-react with GDF11, and vice versa [9,10]. Furthermore, affinity purification from serum using soluble forms of ACVR2A and ACVR2B revealed that they bind not only myostatin, but also GDF11, activins A, B, and AB, BMPs 9, and 10 [11]. Activin type 2 receptors have also been reported to bind inhibins with relatively low affinities compared to activins [96]. Similarly, FST has been shown to bind myostatin, GDF11, activins A, B, AB, and E, inhibins A, and B, BMPs 2, 4, 6, 7, and 15, although the binding affinities for inhibins and BMPs are apparently lower than those for myostatin, GDF11, and activins [12,97]. The fact that several TGF-β family ligands other than myostatin, such as activins [45], also function to suppress muscle growth, which is well demonstrated by substantial increases in muscle mass observed in myostatin null mice after administration of ACVR2B-Fc [37] or transgenic overexpression of FST [40], has promoted enormous interest in developing strategies that target these multiple ligands to more effectively enhance muscle mass. However, unlike myostatin that is primarily expressed in skeletal muscle, activins and BMPs are expressed in a wide variety of tissues to regulate the growth and differentiation of numerous cell types, and therefore, their inhibition may result in serious adverse effects. The potential side effects of myostatin inhibitors will be discussed in the next section.
3. Possible side effects of myostatin inhibitors
Lack of specificity observed in many myostatin inhibitors due to high sequence and structural similarity between mature myostatin and other TGF-β ligands raises the possibility of off-target tissue effects. For instance, clinical development of ACE-031, a soluble ACVR2B receptor designed by Acceleron Pharma for the treatment of DMD, was prematurely terminated due to severe adverse effects including nosebleed, gum bleeding, telangiectasia, and erythema attributed to cross-inhibition of BMP9 and BMP10, ligands involved in endothelial cell function [50]. In addition, a 43% decrease in serum follicle stimulating hormone (FSH), whose synthesis is stimulated by activins, was observed in healthy, postmenopausal women who received a single dose (3 mg/kg) of ACE-031, which caused a near-maximal suppression of activin signaling [80]. Similarly, a significant reduction in FSH was observed in healthy, postmenopausal women when treated with a single dose (1 or 3 mg/kg) of ACE-011, a soluble ACVR2A receptor by Acceleron Pharma. ACE-011, originally designed to treat patients with bone-loss disorders, also resulted in an unexpected increase in the number of RBCs [56], suggested to be caused by inhibition of GDF11 [86,87] although more recent studies have denied the inhibitory action of GDF11 on terminal erythropoiesis [98], leading to its main development direction towards treatment of anemia.
Recently, our group has shown that FST, while significantly enhancing skeletal muscle mass through antagonizing myostatin, exerts negative effects on BMD and induces spontaneous bone fractures likely through blocking GDF11, which we have also demonstrated to play opposite functions to myostatin in regulating bone homeostasis [6]. Intransgenic mice overexpressing FST exhibit decreased quality of skeleton and tibia fractures in contrast to myostatin null mice that display enhanced BMD without fractures (Fig. 2) [6]. Above all, these findings strongly emphasize the importance of target specificity when developing myostatin inhibitors, especially those intended for long-term treatment, to minimize potential adverse effects in different tissues.

Micro-computed tomography images of tibias of 10-week-old wild-type, MSTN-/-, and follistatin-overexpressing (F66) mice. Soft tissue contrast was enhanced using an iodine-based dye. A dramatic increase in muscle mass is observed in both MSTN-/- and F66 mice, but only F66 mice exhibit bone fracture, which is likely induced by inhibition of growth differentiation factor 11.[6]
CONCLUSIONS
The discovery of myostatin as a potent negative regulator of muscle and bone mass immediately generated the possibility that targeting myostatin might be an effective therapeutic strategy to preserve muscle and bone mass in a wide spectrum of conditions associated with debilitating loss of musculoskeletal tissues. Indeed, considerable improvement in understanding of how myostatin activity and signaling is regulated has led numerous pharmaceutical and biotechnology companies to develop promising myostatin inhibitors including antibodies, soluble receptors, and natural antagonists that have successfully progressed through early stages of clinical testing. Clearly, many of these approaches promote robust increases in muscle and bone mass; however, they differ in efficacy and safety. One of the main reasons for this difference lies on the fact that mature myostatin is structurally very similar to other TGF-β family ligands, which may result in cross-reactivity of myostatin antagonists with other closely related growth factors. Importantly, while modulation of multiple factors involved in the myostatin signaling pathway may be more effective in increasing muscle mass than targeting myostatin alone, it can provoke unexpected side effects, as demonstrated by off-target vascular effects caused by ACE-031 through inhibition of BMP 9/10 and bone fractures induced by FST through inhibition of several TGF-β family members including GDF11. Therefore, it is critical to clearly distinguish targets that may potentially enhance the efficacy of a therapeutic agent from those that may cause unwanted adverse effects in order to develop therapeutics that exhibit optimal efficacy with reduced toxicity. Likewise, further research is needed to improve means of achieving higher myostatin specificity, which may provide therapeutic benefits with enhanced safety profiles.
Acknowledgements
This work was supported by Creative-Pioneering Researchers Program through Seoul National University (SNU) and the National Research Foundation of Korea grants (NRF2018R1D1A1B07045334 and NRF-2020R1A2C1010359).
Notes
Ethics approval and consent to participate
Not applicable.
Conflicts of interest
No potential conflict of interest relevant to this article was reported.